

Homologous desensitization of signalling by the alpha (alpha) isoform of the human thromboxane A2 receptor: a specific role for nitric oxide signalling
- PMID: 17466390
- PMCID: PMC2680961
- DOI: 10.1016/j.bbamcr.2007.03.012
Homologous desensitization of signalling by the alpha (alpha) isoform of the human thromboxane A2 receptor: a specific role for nitric oxide signalling
Abstract
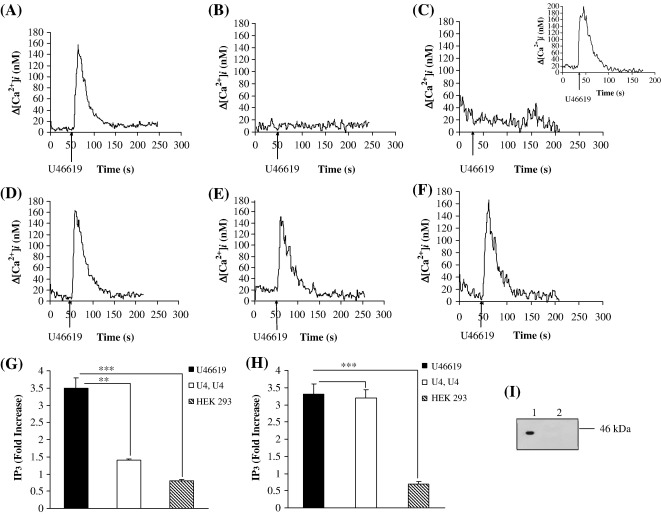
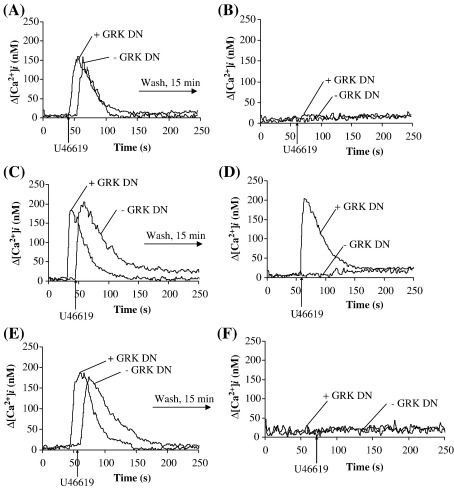
Thromboxane (TX) A(2) plays a central role in hemostasis, regulating platelet activation status and vascular tone. We have recently established that the TP beta isoform of the human TXA(2) receptor (TP) undergoes rapid, agonist-induced homologous desensitization of signalling largely through a G protein-coupled receptor kinase (GRK) 2/3-dependent mechanism with a lesser role for protein kinase (PK) C. Herein, we investigated the mechanism of desensitization of signalling by the TP alpha isoform. TP alpha undergoes profound agonist-induced desensitization of signalling (intracellular calcium mobilization and inositol 1,4,5 trisphosphate generation) in response to the TXA(2) mimetic U46619 but, unlike that of TP beta, this is independent of GRKs. Similar to TP beta, TP alpha undergoes partial agonist-induced desensitization that occurs through a GF 109203X-sensitive, PKC mechanism where Ser(145) within intracellular domain (IC)(2) represents the key phospho-target. TP alpha also undergoes more profound sustained PKC- and PKG-dependent desensitization where Thr(337) and Ser(331), respectively, within its unique C-tail domain were identified as the phospho-targets. Desensitization was impaired by the nitric oxide synthase (NOS), soluble guanylyl cyclase (sGC) and PKG inhibitors L-NAME, LY 83583 and KT5823, respectively, indicating that homologous desensitization of TP alpha involves nitric oxide generation and signalling. Consistent with this, U46619 led to rapid phosphorylation/activation of endogenous eNOS. Collectively, data herein suggest a mechanism whereby agonist-induced PKC phosphorylation of Ser(145) partially and transiently impairs TP alpha signalling while PKG- and PKC-phosphorylation at both Ser(331) and Thr(337), respectively, within its C-tail domain profoundly desensitizes TP alpha, effectively terminating its signalling. Hence, in addition to the agonist-mediated PKC feedback mechanism, U46619-activation of the NOS/sGC/PKG pathway plays a significant role in inducing homologous desensitization of TP alpha.
Figures








Similar articles
-
Homologous desensitization of signalling by the beta (beta) isoform of the human thromboxane A2 receptor.Biochim Biophys Acta. 2006 Sep;1761(9):1114-31. doi: 10.1016/j.bbalip.2006.07.012. Epub 2006 Aug 3. Biochim Biophys Acta. 2006. PMID: 16956790
-
The alpha, but not the beta, isoform of the human thromboxane A2 receptor is a target for nitric oxide-mediated desensitization. Independent modulation of Tp alpha signaling by nitric oxide and prostacyclin.J Biol Chem. 2003 Dec 19;278(51):51190-202. doi: 10.1074/jbc.M309314200. Epub 2003 Oct 6. J Biol Chem. 2003. Retraction in: J Biol Chem. 2016 Sep 9;291(37):19260. doi: 10.1074/jbc.A116.309314. PMID: 14530262 Retracted.
-
Prostaglandin D(2) receptor-mediated desensitization of the alpha isoform of the human thromboxane A(2) receptor.Biochem Pharmacol. 2001 Jul 15;62(2):229-39. doi: 10.1016/s0006-2952(01)00661-x. Biochem Pharmacol. 2001. PMID: 11389883
-
Cell signalling through thromboxane A2 receptors.Cell Signal. 2004 May;16(5):521-33. doi: 10.1016/j.cellsig.2003.10.008. Cell Signal. 2004. PMID: 14751539 Review.
-
IRAG and novel PKG targeting in the cardiovascular system.Am J Physiol Heart Circ Physiol. 2011 Sep;301(3):H672-82. doi: 10.1152/ajpheart.00198.2011. Epub 2011 Jun 10. Am J Physiol Heart Circ Physiol. 2011. PMID: 21666108 Review.
Cited by
-
Thromboxane-induced contractile response of mesenteric arterioles is diminished in the older rats and the older hypertensive rats.Front Pharmacol. 2022 Oct 12;13:1019511. doi: 10.3389/fphar.2022.1019511. eCollection 2022. Front Pharmacol. 2022. PMID: 36313372 Free PMC article.
-
The Wilms' tumour suppressor protein WT1 acts as a key transcriptional repressor of the human thromboxane A2 receptor gene in megakaryocytes.J Cell Mol Med. 2009 Nov-Dec;13(11-12):4571-86. doi: 10.1111/j.1582-4934.2008.00599.x. J Cell Mol Med. 2009. PMID: 19067769 Free PMC article.
-
Protein kinase C-related kinase 1 and 2 play an essential role in thromboxane-mediated neoplastic responses in prostate cancer.Oncotarget. 2015 Sep 22;6(28):26437-56. doi: 10.18632/oncotarget.4664. Oncotarget. 2015. PMID: 26296974 Free PMC article.
-
Role of GRK6 in the Regulation of Platelet Activation through Selective G Protein-Coupled Receptor (GPCR) Desensitization.Int J Mol Sci. 2020 May 30;21(11):3932. doi: 10.3390/ijms21113932. Int J Mol Sci. 2020. PMID: 32486261 Free PMC article.
-
A Review of Prostanoid Receptors: Expression, Characterization, Regulation, and Mechanism of Action.J Cell Commun Signal. 2021 Jun;15(2):155-184. doi: 10.1007/s12079-020-00585-0. Epub 2020 Sep 24. J Cell Commun Signal. 2021. PMID: 32970276 Free PMC article. Review.
References
-
- Narumiya S., Sugimoto Y., Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol. Rev. 1999;79:1193–1226. - PubMed
-
- Sugimoto Y. Physiological functions of prostanoid receptors and their subtypes. Nippon Yakurigaku Zasshi. 2000;115:131–141. - PubMed
-
- Hanasaki K., Nakano T., Arita H. Receptor-mediated mitogenic effect of thromboxane A2 in vascular smooth muscle cells. Biochem. Pharmacol. 1990;40:2535–2542. - PubMed
-
- Dorn G.W., II, Becker M.W., Davis M.G. Dissociation of the contractile and hypertrophic effects of vasoconstrictor prostanoids in vascular smooth muscle. J. Biol. Chem. 1992;267:24897–24905. - PubMed
-
- Ali S., Davis M.G., Becker M.W., Dorn G.W., II Thromboxane A2 stimulates vascular smooth muscle hypertrophy by up-regulating the synthesis and release of endogenous basic fibroblast growth factor. J. Biol. Chem. 1993;268:17397–17403. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
