

Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis
- PMID: 17470635
- PMCID: PMC2064806
- DOI: 10.1083/jcb.200610086
Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis
Abstract
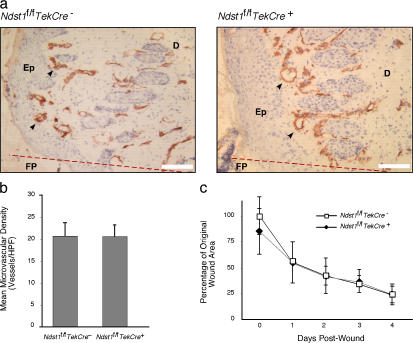
To examine the role of endothelial heparan sulfate during angiogenesis, we generated mice bearing an endothelial-targeted deletion in the biosynthetic enzyme N-acetylglucosamine N-deacetylase/N-sulfotransferase 1 (Ndst1). Physiological angiogenesis during cutaneous wound repair was unaffected, as was growth and reproductive capacity of the mice. In contrast, pathological angiogenesis in experimental tumors was altered, resulting in smaller tumors and reduced microvascular density and branching. To simulate the angiogenic environment of the tumor, endothelial cells were isolated and propagated in vitro with proangiogenic growth factors. Binding of FGF-2 and VEGF(164) to cells and to purified heparan sulfate was dramatically reduced. Mutant endothelial cells also exhibited altered sprouting responses to FGF-2 and VEGF(164), reduced Erk phosphorylation, and an increase in apoptosis in branching assays. Corresponding changes in growth factor binding to tumor endothelium and apoptosis were also observed in vivo. These findings demonstrate a cell-autonomous effect of heparan sulfate on endothelial cell growth in the context of tumor angiogenesis.
Figures






References
-
- Bai, X., G. Wei, A. Sinha, and J.D. Esko. 1999. Chinese hamster ovary cell mutants defective in glycosaminoglycan assembly and glucuronosyltransferase I. J. Biol. Chem. 274:13017–13024. - PubMed
-
- Bame, K.J., and J.D. Esko. 1989. Undersulfated heparan sulfate in a Chinese hamster ovary cell mutant defective in heparan sulfate N-sulfotransferase. J. Biol. Chem. 264:8059–8065. - PubMed
-
- Bergsland, E.K. 2004. Update on clinical trials targeting vascular endothelial growth factor in cancer. Am. J. Health Syst. Pharm. 61:S12–S20. - PubMed
-
- Bix, G., and R.V. Iozzo. 2005. Matrix revolutions: “tails” of basement-membrane components with angiostatic functions. Trends Cell Biol. 15:52–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
