

Structure of the Q67H mutant of R67 dihydrofolate reductase-NADP+ complex reveals a novel cofactor binding mode
- PMID: 17473013
- PMCID: PMC2206676
- DOI: 10.1110/ps.062740907
Structure of the Q67H mutant of R67 dihydrofolate reductase-NADP+ complex reveals a novel cofactor binding mode
Abstract
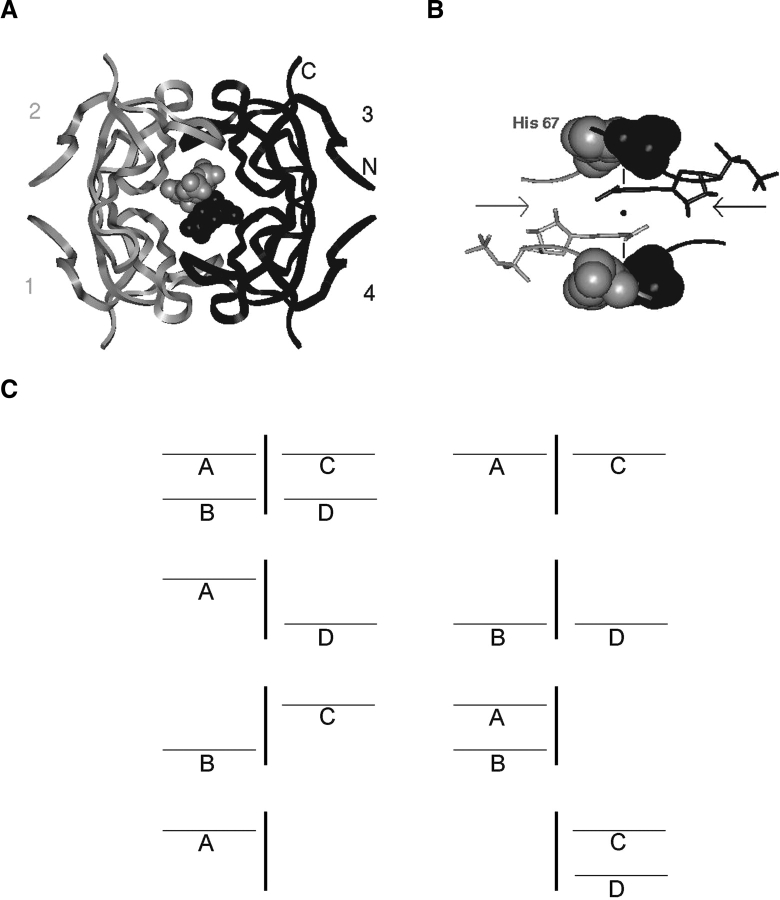
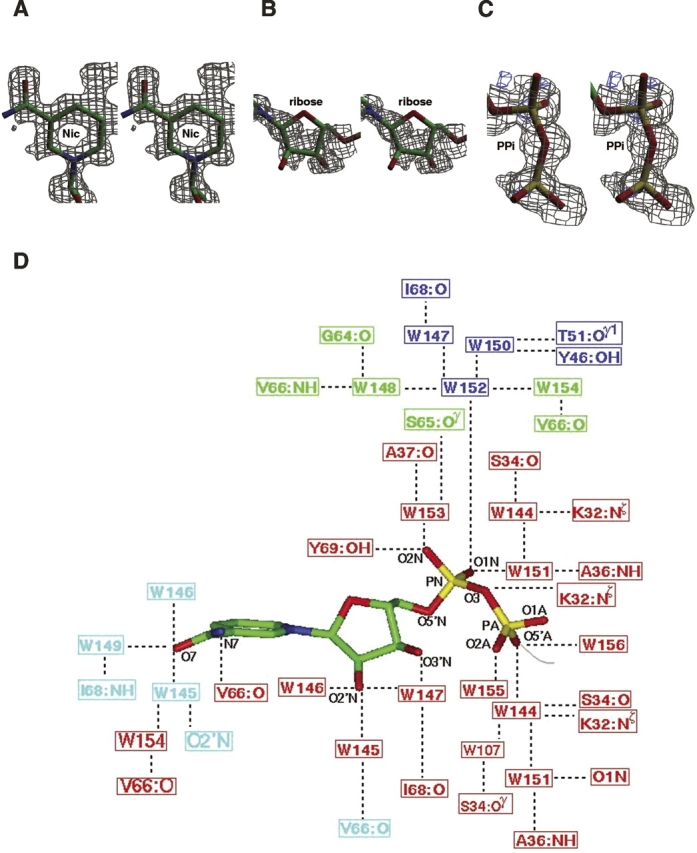
Plasmid-encoded bacterial R67 dihydrofolate reductase (DHFR) is a NADPH-dependent enzyme unrelated to chromosomal DHFR in amino acid sequence and structure. R67 DHFR is insensitive to the bacterial drug trimethoprim in contrast to chromosomal DHFR. The crystal structure of Q67H mutant of R67 DHFR bound to NADP(+) has been determined at 1.15 angstroms resolution. The cofactor assumes an extended conformation with the nicotinamide ring bound near the center of the active site pore, the ribose and pyrophosphate group (PP(i)) extending toward the outer pore. The ribonicotinamide exhibits anti conformation as in chromosomal DHFR complexes. The relative orientation between the PP(i) and the nicotinamide ribose differs from that observed in chromosomal DHFR-NADP(+) complexes. The coenzyme displays symmetrical binding mode with several water-mediated hydrogen bonds with the protein besides ionic, stacking, and van der Waals interactions. The structure provides a molecular basis for the observed stoichiometry and cooperativity in ligand binding. The ternary model based on the present structure and the previous R67 DHFR-folate complex provides insight into the catalytic mechanism and indicates that the relative orientation of the reactants in plasmid DHFR is different from that seen in chromosomal DHFRs.
Figures
Similar articles
-
A glutamine 67--> histidine mutation in homotetrameric R67 dihydrofolate reductase results in four mutations per single active site pore and causes substantial substrate and cofactor inhibition.Protein Eng. 1997 Dec;10(12):1415-24. doi: 10.1093/protein/10.12.1415. Protein Eng. 1997. PMID: 9543003
-
Unusual binding stoichiometries and cooperativity are observed during binary and ternary complex formation in the single active pore of R67 dihydrofolate reductase, a D2 symmetric protein.Biochemistry. 1996 Sep 3;35(35):11414-24. doi: 10.1021/bi960205d. Biochemistry. 1996. PMID: 8784197
-
Interligand Overhauser effects in type II dihydrofolate reductase.Biochemistry. 2001 Apr 10;40(14):4242-52. doi: 10.1021/bi0026425. Biochemistry. 2001. PMID: 11284680
-
Searching sequence space: two different approaches to dihydrofolate reductase catalysis.Chembiochem. 2005 Apr;6(4):590-600. doi: 10.1002/cbic.200400237. Chembiochem. 2005. PMID: 15812782 Review.
-
Basis of selectivity of antibacterial diaminopyrimidines.J Chemother. 1993 Dec;5(6):393-9. J Chemother. 1993. PMID: 8195830 Review.
Cited by
-
Novel crystallization conditions for tandem variant R67 DHFR yield a wild-type crystal structure.Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011 Nov 1;67(Pt 11):1316-22. doi: 10.1107/S1744309111030417. Epub 2011 Oct 25. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011. PMID: 22102224 Free PMC article.
-
Crystal structure of the plasmid-encoded R67 dihydrofolate reductase complexed with Congo red an amyloid binding dye.Sci Rep. 2025 Feb 12;15(1):5212. doi: 10.1038/s41598-025-89539-3. Sci Rep. 2025. PMID: 39939735 Free PMC article.
-
Crystal structure of a type II dihydrofolate reductase catalytic ternary complex.Biochemistry. 2007 Dec 25;46(51):14878-88. doi: 10.1021/bi701532r. Epub 2007 Dec 4. Biochemistry. 2007. PMID: 18052202 Free PMC article.
References
-
- Andres J., Moliner, V., Safont, V.S., Domingo, L.R., Picher, M.T., and Krechl, J. 1996. On transition structures for hydride transfer step: A theoretical study of the reaction catalyzed by dihydrofolate reductase enzyme. Bioorg. Chem. 24: 10–18.
-
- Bellamacina C.R. 1996. The nicotinamide dinucleotide binding motif: A comparison of nucleotide binding proteins. FASEB J. 10: 1257–1269. - PubMed
-
- Bradrick T.D., Beechem, J.M., and Howell, E.E. 1996. Unusual binding stoichiometries and cooperativity are observed during binary and ternary complex formation in the single active pore of R67 dihydrofolate reductase, a D2 symmetric protein. Biochemistry 35: 11414–11424. - PubMed
-
- Brito R.M., Rudolph, F.B., and Rosevear, P.R. 1991. Conformation of NADP+ bound to a type II dihydrofolate reductase. Biochemistry 30: 1461–1469. - PubMed
-
- Brunger A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. - PubMed
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Miscellaneous
