

Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action
- PMID: 17476361
- PMCID: PMC1857259
- DOI: 10.1172/JCI30558
Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action
Abstract
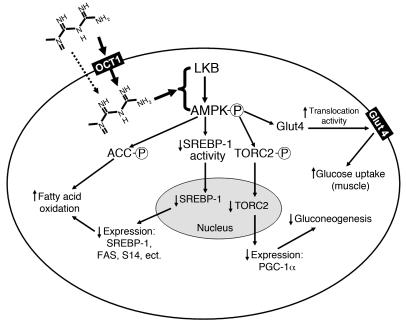
Metformin is among the most widely prescribed drugs for the treatment of type 2 diabetes. Organic cation transporter 1 (OCT1) plays a role in the hepatic uptake of metformin, but its role in the therapeutic effects of the drug, which involve activation of AMP-activated protein kinase (AMPK), is unknown. Recent studies have shown that human OCT1 is highly polymorphic. We investigated whether OCT1 plays a role in the action of metformin and whether individuals with OCT1 polymorphisms have reduced response to the drug. In mouse hepatocytes, deletion of Oct1 resulted in a reduction in the effects of metformin on AMPK phosphorylation and gluconeogenesis. In Oct1-deficient mice the glucose-lowering effects of metformin were completely abolished. Seven nonsynonymous polymorphisms of OCT1 that exhibited reduced uptake of metformin were identified. Notably, OCT1-420del (allele frequency of about 20% in white Americans), previously shown to have normal activity for model substrates, had reduced activity for metformin. In clinical studies, the effects of metformin in glucose tolerance tests were significantly lower in individuals carrying reduced function polymorphisms of OCT1. Collectively, the data indicate that OCT1 is important for metformin therapeutic action and that genetic variation in OCT1 may contribute to variation in response to the drug.
Figures
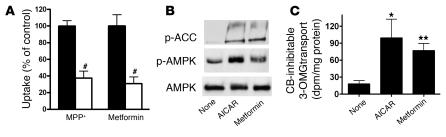
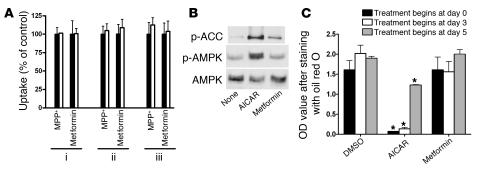
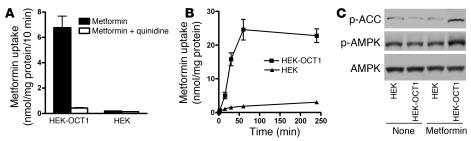
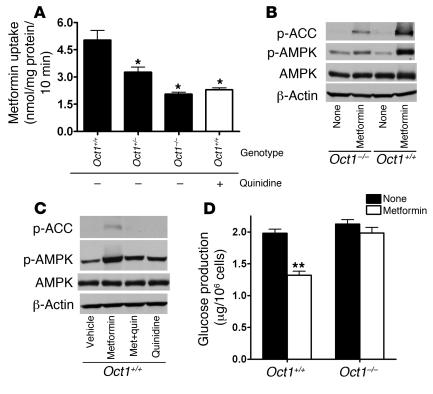
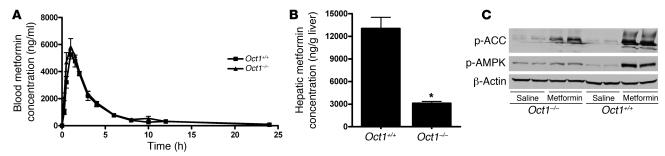
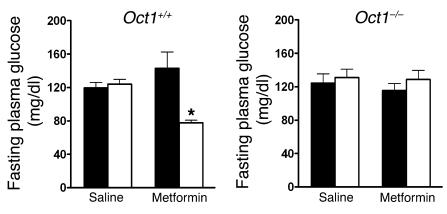
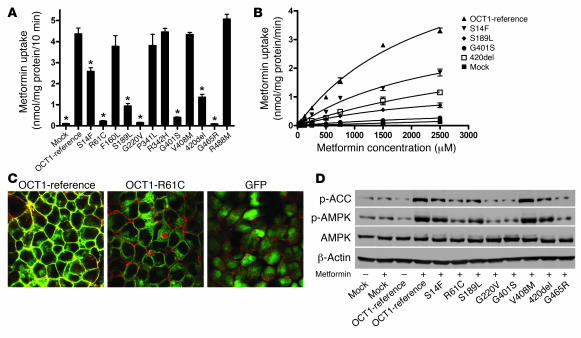
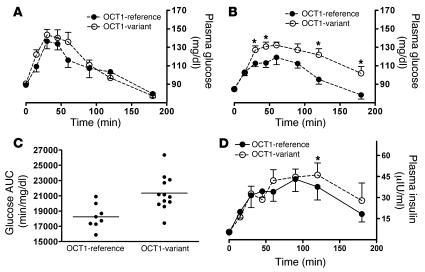
Comment in
-
Pharmacogenetics of metformin response: a step in the path toward personalized medicine.J Clin Invest. 2007 May;117(5):1226-9. doi: 10.1172/JCI32133. J Clin Invest. 2007. PMID: 17476355 Free PMC article. Review.
References
-
- Kirpichnikov D., McFarlane S.I., Sowers J.R. Metformin: an update. Ann. Intern. Med. 2002;137:25–33. - PubMed
-
- Lin H.Z., et al. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat. Med. 2000;6:998–1003. - PubMed
-
- Baillargeon J.P., Iuorno M.J., Nestler J.E. Insulin sensitizers for polycystic ovary syndrome. Clin. Obstet. Gynecol. 2003;46:325–340. - PubMed
-
- Abbud W., et al. Stimulation of AMP-activated protein kinase (AMPK) is associated with enhancement of Glut1-mediated glucose transport. Arch. Biochem. Biophys. 2000;380:347–352. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
