

Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia
- PMID: 17478498
- PMCID: PMC1904289
- DOI: 10.1093/nar/gkm271
Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia
Abstract
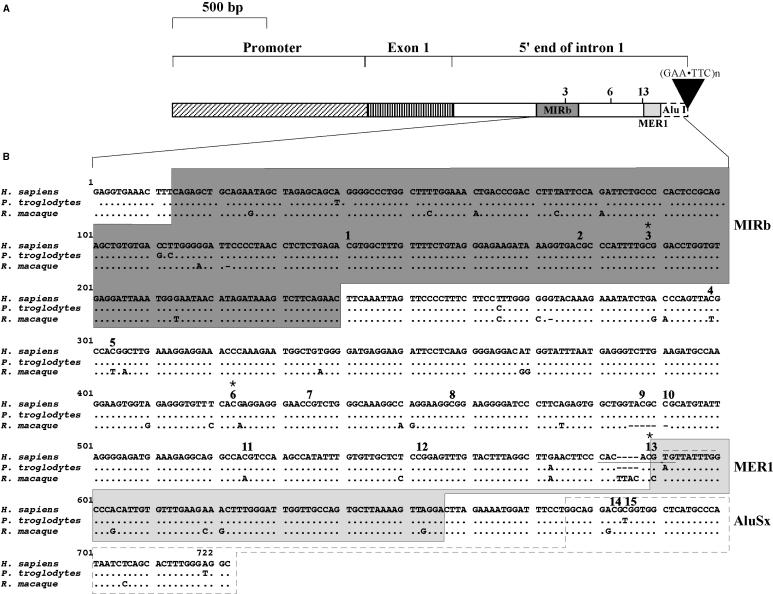
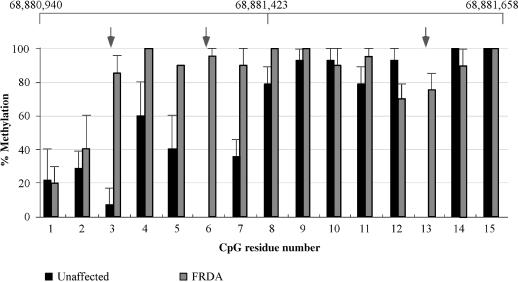
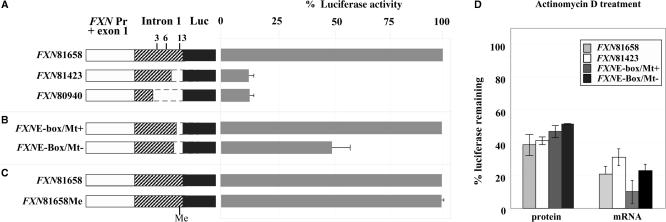
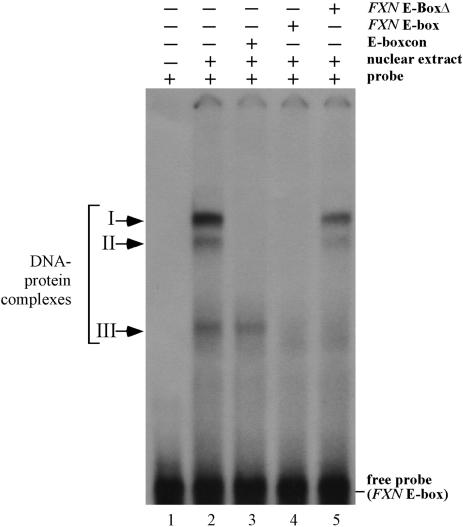
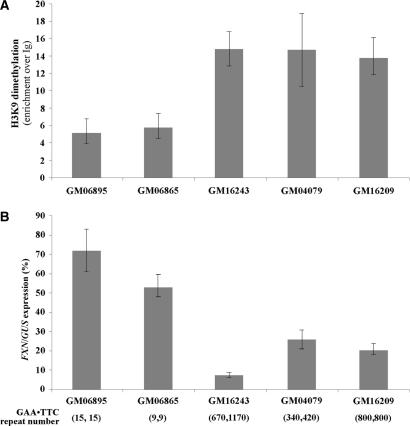
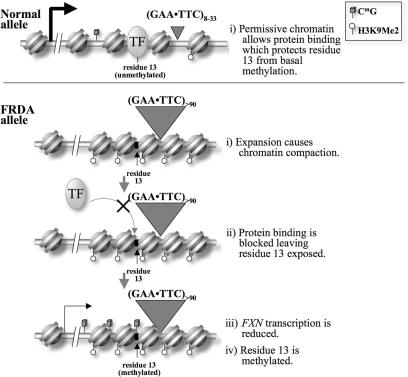
Friedreich ataxia (FRDA), the most common hereditary ataxia, is caused by mutations in the frataxin (FXN) gene. The vast majority of FRDA mutations involve expansion of a GAA*TTC-repeat tract in intron 1, which leads to an FXN mRNA deficit. Bisulfite mapping demonstrates that the region adjacent to the repeat was methylated in both unaffected and affected individuals. However, methylation was more extensive in patients. Additionally, three residues were almost completely methylation-free in unaffected individuals but almost always methylated in those with FRDA. One of these residues is located within an E-box whose deletion caused a significant drop in promoter activity in reporter assays. Elevated levels of histone H3 dimethylated on lysine 9 were seen in FRDA cells consistent with a more repressive chromatin organization. Such chromatin is known to reduce transcription elongation. This may be one way in which the expanded repeats contribute to the frataxin deficit in FRDA. Our data also suggest that repeat-mediated chromatin changes may also affect transcription initiation by blocking binding of factors that increase frataxin promoter activity. Our results also raise the possibility that the repeat-mediated increases in DNA methylation in the FXN gene in FRDA patients are secondary to the chromatin changes.
Figures
Similar articles
-
Long intronic GAA*TTC repeats induce epigenetic changes and reporter gene silencing in a molecular model of Friedreich ataxia.Nucleic Acids Res. 2008 Nov;36(19):6056-65. doi: 10.1093/nar/gkn604. Epub 2008 Sep 27. Nucleic Acids Res. 2008. PMID: 18820300 Free PMC article.
-
Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells.J Biol Chem. 2011 Feb 11;286(6):4209-15. doi: 10.1074/jbc.M110.194035. Epub 2010 Dec 2. J Biol Chem. 2011. PMID: 21127046 Free PMC article.
-
The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues.Hum Mol Genet. 2008 Mar 1;17(5):735-46. doi: 10.1093/hmg/ddm346. Epub 2007 Nov 27. Hum Mol Genet. 2008. PMID: 18045775
-
Beyond loss of frataxin: the complex molecular pathology of Friedreich ataxia.Discov Med. 2014 Jan;17(91):25-35. Discov Med. 2014. PMID: 24411698 Review.
-
Analysis of Putative Epigenetic Regulatory Elements in the FXN Genomic Locus.Int J Mol Sci. 2020 May 12;21(10):3410. doi: 10.3390/ijms21103410. Int J Mol Sci. 2020. PMID: 32408537 Free PMC article. Review.
Cited by
-
Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich's ataxia.Nucleic Acids Res. 2011 Oct;39(19):8366-77. doi: 10.1093/nar/gkr542. Epub 2011 Jul 10. Nucleic Acids Res. 2011. PMID: 21745819 Free PMC article.
-
DNA dynamics is likely to be a factor in the genomic nucleotide repeats expansions related to diseases.PLoS One. 2011;6(5):e19800. doi: 10.1371/journal.pone.0019800. Epub 2011 May 20. PLoS One. 2011. PMID: 21625483 Free PMC article.
-
Long range regulation of human FXN gene expression.PLoS One. 2011;6(7):e22001. doi: 10.1371/journal.pone.0022001. Epub 2011 Jul 8. PLoS One. 2011. PMID: 21760943 Free PMC article.
-
Identification of restriction endonucleases sensitive to 5-cytosine methylation at non-CpG sites, including expanded (CAG)n/(CTG)n repeats.Epigenetics. 2011 Apr;6(4):416-20. doi: 10.4161/epi.6.4.14953. Epub 2011 Apr 1. Epigenetics. 2011. PMID: 21364324 Free PMC article.
-
Long intronic GAA*TTC repeats induce epigenetic changes and reporter gene silencing in a molecular model of Friedreich ataxia.Nucleic Acids Res. 2008 Nov;36(19):6056-65. doi: 10.1093/nar/gkn604. Epub 2008 Sep 27. Nucleic Acids Res. 2008. PMID: 18820300 Free PMC article.
References
-
- Pandolfo M. The molecular basis of Friedreich ataxia. Adv. Exp. Med. Biol. 2002;516:99–118. - PubMed
-
- Pianese L, Turano M, Lo Casale MS, De Biase I, Giacchetti M, Monticelli A, Criscuolo C, Filla A, Cocozza S. Real time PCR quantification of frataxin mRNA in the peripheral blood leucocytes of Friedreich ataxia patients and carriers. J. Neurol. Neurosurg. Psychiatry. 2004;75:1061–1063. - PMC - PubMed
-
- Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
