

Trypanosoma brucei homologous recombination is dependent on substrate length and homology, though displays a differential dependence on mismatch repair as substrate length decreases
- PMID: 17478508
- PMCID: PMC1904282
- DOI: 10.1093/nar/gkm249
Trypanosoma brucei homologous recombination is dependent on substrate length and homology, though displays a differential dependence on mismatch repair as substrate length decreases
Abstract
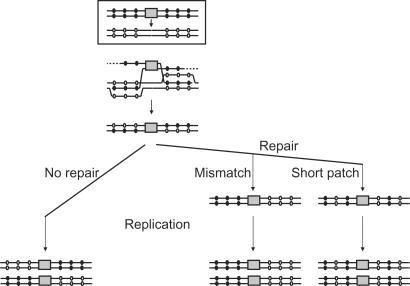
Homologous recombination functions universally in the maintenance of genome stability through the repair of DNA breaks and in ensuring the completion of replication. In some organisms, homologous recombination can perform more specific functions. One example of this is in antigenic variation, a widely conserved mechanism for the evasion of host immunity. Trypanosoma brucei, the causative agent of sleeping sickness in Africa, undergoes antigenic variation by periodic changes in its variant surface glycoprotein (VSG) coat. VSG switches involve the activation of VSG genes, from an enormous silent archive, by recombination into specialized expression sites. These reactions involve homologous recombination, though they are characterized by an unusually high rate of switching and by atypical substrate requirements. Here, we have examined the substrate parameters of T. brucei homologous recombination. We show, first, that the reaction is strictly dependent on substrate length and that it is impeded by base mismatches, features shared by homologous recombination in all organisms characterized. Second, we identify a pathway of homologous recombination that acts preferentially on short substrates and is impeded to a lesser extent by base mismatches and the mismatch repair machinery. Finally, we show that mismatches during T. brucei recombination may be repaired by short-patch mismatch repair.
Figures
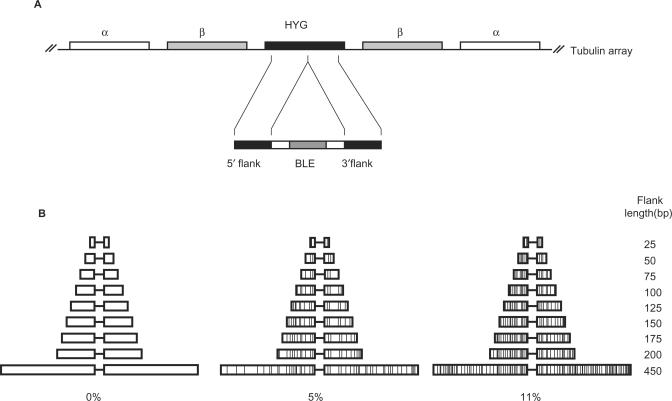
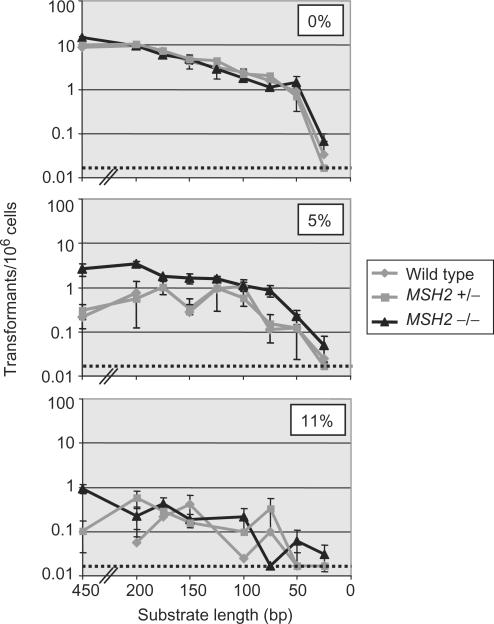
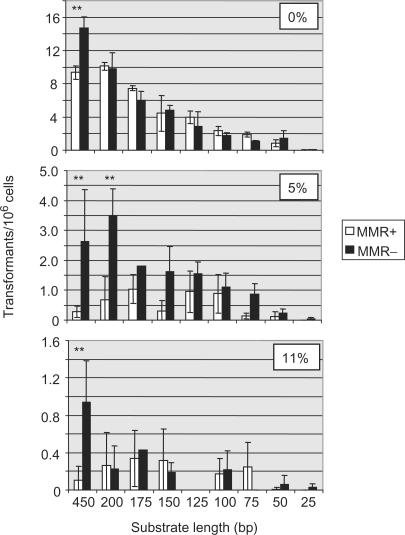
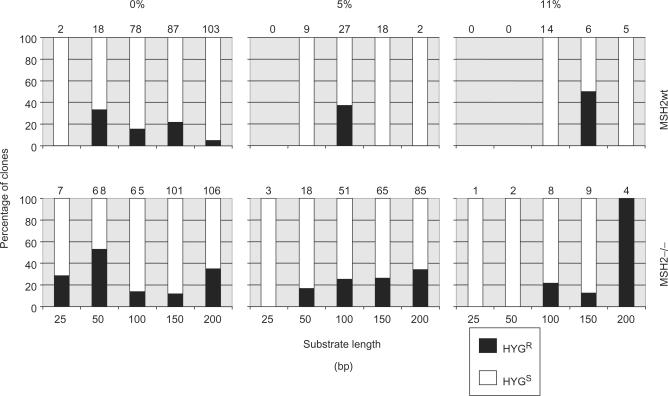
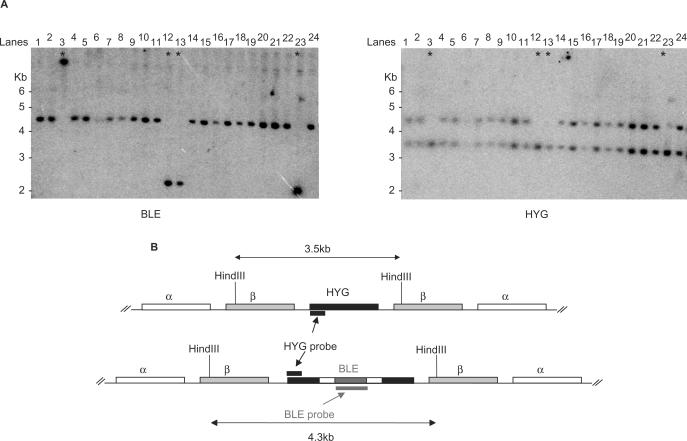
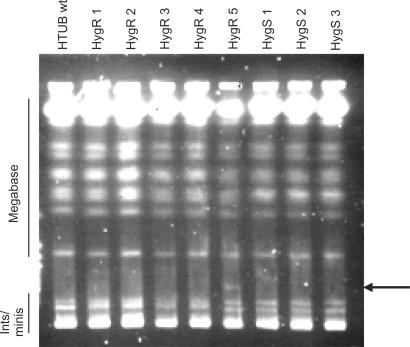
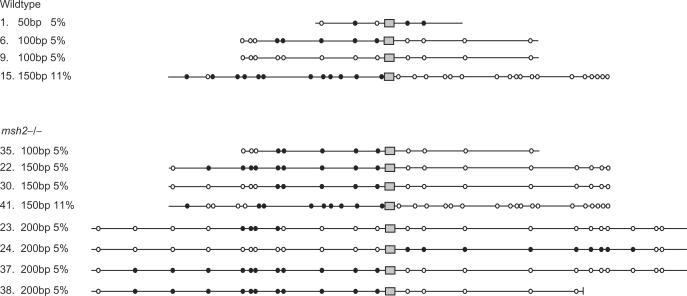
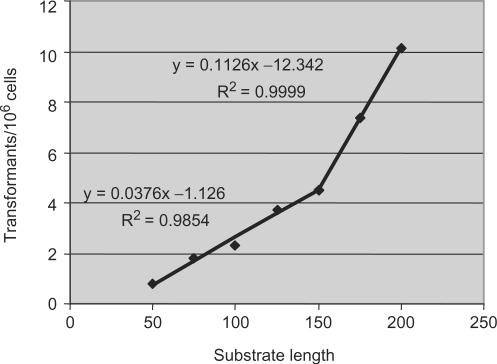
Similar articles
-
Mismatch repair regulates homologous recombination, but has little influence on antigenic variation, in Trypanosoma brucei.J Biol Chem. 2003 Nov 14;278(46):45182-8. doi: 10.1074/jbc.M308123200. Epub 2003 Aug 21. J Biol Chem. 2003. PMID: 12933800
-
Trypanosoma brucei BRCA2 acts in antigenic variation and has undergone a recent expansion in BRC repeat number that is important during homologous recombination.Mol Microbiol. 2008 Jun;68(5):1237-51. doi: 10.1111/j.1365-2958.2008.06230.x. Epub 2008 Apr 22. Mol Microbiol. 2008. PMID: 18430140 Free PMC article.
-
Two pathways of homologous recombination in Trypanosoma brucei.Mol Microbiol. 2002 Sep;45(6):1687-700. doi: 10.1046/j.1365-2958.2002.03122.x. Mol Microbiol. 2002. PMID: 12354234
-
Escaping the immune system by DNA repair and recombination in African trypanosomes.Open Biol. 2019 Nov 29;9(11):190182. doi: 10.1098/rsob.190182. Epub 2019 Nov 13. Open Biol. 2019. PMID: 31718509 Free PMC article. Review.
-
DNA Recombination Strategies During Antigenic Variation in the African Trypanosome.Microbiol Spectr. 2015 Apr;3(2):MDNA3-0016-2014. doi: 10.1128/microbiolspec.MDNA3-0016-2014. Microbiol Spectr. 2015. PMID: 26104717 Review.
Cited by
-
DNA double-strand breaks and telomeres play important roles in trypanosoma brucei antigenic variation.Eukaryot Cell. 2015 Mar;14(3):196-205. doi: 10.1128/EC.00207-14. Epub 2015 Jan 9. Eukaryot Cell. 2015. PMID: 25576484 Free PMC article. Review.
-
Gene conversion transfers the GAF-A domain of phosphodiesterase TbrPDEB1 to one allele of TbrPDEB2 of Trypanosoma brucei.PLoS Negl Trop Dis. 2009 Jun 9;3(6):e455. doi: 10.1371/journal.pntd.0000455. PLoS Negl Trop Dis. 2009. PMID: 19513125 Free PMC article.
-
Interactions among Trypanosoma brucei RAD51 paralogues in DNA repair and antigenic variation.Mol Microbiol. 2011 Jul;81(2):434-56. doi: 10.1111/j.1365-2958.2011.07703.x. Epub 2011 May 26. Mol Microbiol. 2011. PMID: 21615552 Free PMC article.
-
A toolkit enabling efficient, scalable and reproducible gene tagging in trypanosomatids.Open Biol. 2015 Jan;5(1):140197. doi: 10.1098/rsob.140197. Open Biol. 2015. PMID: 25567099 Free PMC article.
-
Antigenic variation and transmission fitness as drivers of bacterial strain structure.Cell Microbiol. 2013 Dec;15(12):1969-75. doi: 10.1111/cmi.12182. Epub 2013 Aug 28. Cell Microbiol. 2013. PMID: 23941262 Free PMC article. Review.
References
-
- West SC. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 2003;4:435–445. - PubMed
-
- Story RM, Weber IT, Steitz TA. The structure of the E. coli recA protein monomer and polymer. Nature. 1992;355:318–325. - PubMed
-
- Conway AB, Lynch TW, Zhang Y, Fortin GS, Fung CW, Symington LS, Rice PA. Crystal structure of a Rad51 filament. Nat. Struct. Mol. Biol. 2004;11:791–796. - PubMed
