

Review
doi: 10.1016/j.cytogfr.2007.04.006.
Epub 2007 May 7.
Autophagy and NF-kappaB: fight for fate
Affiliations
- PMID: 17485237
- PMCID: PMC2810660
- DOI: 10.1016/j.cytogfr.2007.04.006
Item in Clipboard
Review
Autophagy and NF-kappaB: fight for fate
Cytokine Growth Factor Rev.
2007 Jun-Aug.
Abstract
Autophagy/macroautophagy is known for its role in cellular homeostasis, development, cell survival, aging, immunity, cancer and neurodegeneration. However, until recently, the mechanisms by which autophagy contributes to these important processes were largely unknown. The demonstration of a direct cross-talk between autophagy and NF-kappaB opens up new frontiers for deciphering the role of autophagy in diverse biological processes. Here, we review our current understanding of autophagy, with a focus on its role in tumor suppression, NF-kappaB inactivation and selective protein degradation in mammals. We also list some most intriguing and outstanding questions that are likely to engage researchers in the near future.
Figures
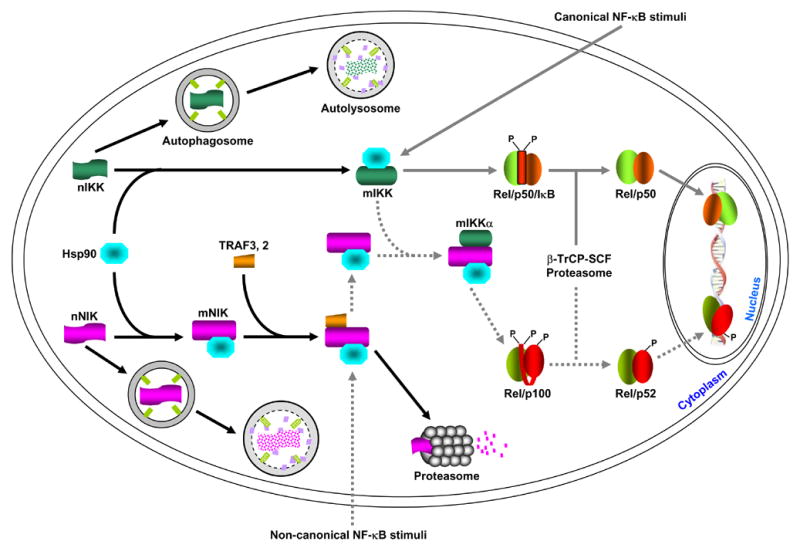
Newly synthesized IKK and NIK proteins (labeled as nIKK and nNIK, respectively) are rapidly captured by the Hsp90 chaperone complex (for simplicity, the three IKK subunits, IKKα, IKKβ and IKKγ, are just indicated as IKK). Two possible roles of this association are: promote the maturation and/or maintain the correct conformation of the IKK and NIK proteins. When Hsp90 function is absence (e.g. inhibition by geldanamycin, GA), the nascent IKK and NIK proteins cannot maturated and/or the mature proteins (labeled as mIKK and mNIK) cannot maintain the correct conformation, resulting in degradation via the autophagy pathway. In response to the classical NF-κB stimuli, IKK is activated, therefore leading to IκB proteasomal degradation and NF-κB activation. Hsp90 seems also involved in the formation of a large IKK signalsome required for IKK activation, although it is not required for the association among the three IKK subunits themselves. Unlike the mature IKK proteins that are relatively stable, the mature NIK proteins are quickly degraded by the proteasome. This proteasomal degradation is mediated by TRAF3 and possibly also by other proteins such as TRAF2. The non-canonical NF-κB stimuli somehow lead to TRAF3 degradation and/or dissociation from NIK, thereby protecting them from the proteasomal degradation. The NIK proteins freed from TRAF3 then have a chance to activate and recruit the mature IKKα into p100 for its processing and subsequent NF-κB activation. Although it always binds to NIK, Hsp90 is not required for the formation of the NIK/IKKα/p100 complex. However, it remains to be investigated whether Hsp90 is involved in the NIK regulation by TRAF3 or by the upstream NF-κB stimuli. Of note, IKK and NIK proteins escaped from GA-mediated autophagic degradation by autophagy inhibition remain their abilities, at least partially, in NF-κB activation. These results suggested that nascent IKK and NIK proteins may have some activity or they can become mature, possibly less efficiently, without Hsp90. So, the major role of Hsp90 might be to suppress autophagic degradation of IKK and NIK but not to help their folding.
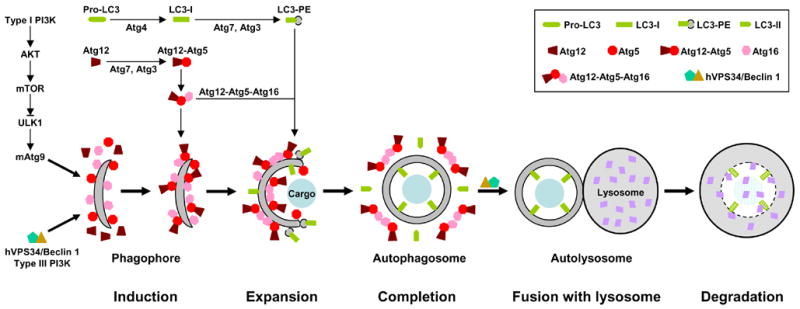
Autophagy involves at least five steps: 1. induction; 2. expansion; 3. completion; 4. docking and fusion; 5. degradation. For example, nutrient starvation, the prototypic stimulus of autophagy, leads to inhibition of the type I PI3K-AKT-mTOR signaling and activation of type III PI3K hVPS34/Beclin 1 (Atg6). Inhibition of mTOR unleashes ULK1 (Atg1), which in turn induces redistribution of mAtg9 from trans-Golgi to late endosome. Activation of hVPS34/Beclin 1, on the other hand, generates PIP3 [PtdIns(3,4,5)P3] on endomembrane. These events result in isolation and decoration with Atg5 and Atg16 of a small template membrane dubbed as phagophore. The expansion of this crescent structure requires conjugation of two ubiquitin-like proteins Atg12 and LC3 (Atg8) to Atg5 and phosphatidylethanolamine (PE), respectively. The Atg12-Atg5 conjugate binds to Atg16 and this trimeric complex polymerizes. The Atg12-Atg5-Atg16 oligomers on the surface, particularly at the tips, of phagophore initiate its elongation and curvature by recruiting cytosolic LC3-PE. The cargo may also contribute to the initiation and expansion of phagophore by serving as a scaffold. During the formation of autophagosome, the Atg12-Atg5-Atg16 complexes redistribute and concentrate mostly on the external lipid bilayers. Once the autophagosome is completed, the Atg12-Atg5-Atg16 complexes dissociate from it. The LC-II on the external lipid bilayer of the autophagosome is also released into the cytosol due to the cleavage mediated by Atg4. The uncoated autophagosome then fuses with the lysosome with the help of hVPS34/Beclin 1. The sequestered cargo together with the LC-II trapped in the lumen of the autophagosome is degraded within the autolysosome. During autophagic process LC3 is converted from LC-I (non-lipidated, long form) to LC3-II (PE-conjugated, short form) and translocated sequentially from the cytosol to phagophores, autophagosomes and autolysosome (punctate structures), it can be monitored easily by immunoblotting and immunofluresecnce/fluresecence, so it is the best and most used marker for autophagy. Currently, 3-MA (3-methyladenine) and AICAR (5-aminoimidazole-4-carboxaminde riboside) are two most widely used inhibitors for autophagy. The inhibitory function of 3-MA is attributed to its ability in suppressing type III PI3K, although it also suppresses type I PI3K. On the other hand, the mechanism by which AICAR inhibits autophagy is still unclear. One possibility is that AICAR activates p70S6K (70 kDa kinase for ribosomal protein S6) by functioning as an activator of AMPK (AMP-activated protein kinase), the original function of AICAR, although activated AMPK can inhibit mTOR. Other possibilities are that AICAR may inhibit type III PI3K (similar to 3-MA) or it may actually activate mTOR via an unidentified mechanism (35–37, also see Figure 3).
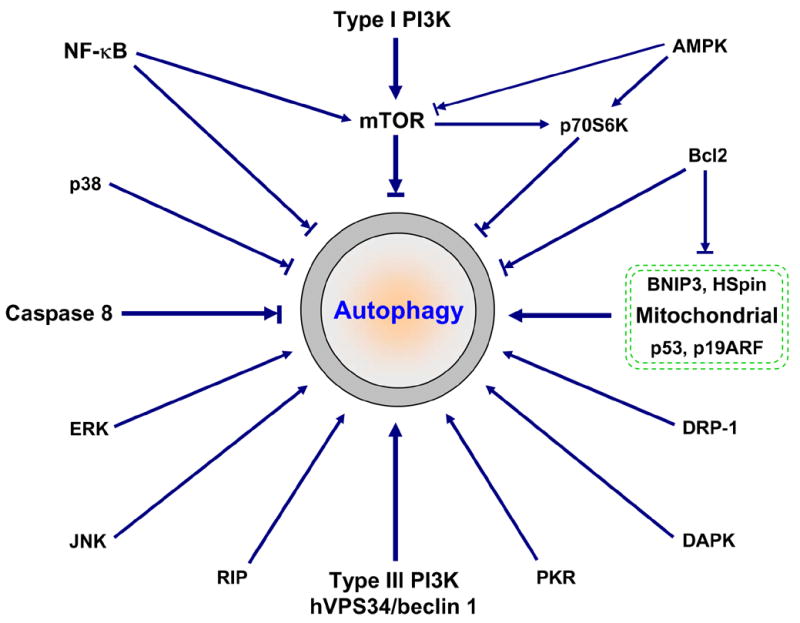
In addition to type I and type III PI3K signaling (Figure 2), many other signaling proteins have been linked to autophagy regulation. These signaling proteins often cross-talk and form a very complicated regulatory net. For example, NF-κB-mediated autophagy repression may involve mTOR activation, Bcl2/Bcl-xL upregualtion and RIP degradation (6, 38). Upregulated Bcl2/Bcl-xL can bind to and suppress the inhibitory function on autophagy of BNIP3 and HSpin, two BH-3-like domain containing protein located at mitochondrial (39, 40). Bcl2 may also bind to Beclin 1, thereby directly regulating autophagy (41, 42). Similar to NF-κB activation, activated caspase 8 cleaves RIP, leading to suppression of autophagy. There are at least two different but related mechanisms involved in RIP-mediated activation of autophagy: JNK activation and mitochondria stimuli (ROS accumulation, ATP depletion and Ca2+ release, etc.) (see Figure 5 for details). Tumor suppressors p53 (via DRAM) and p19ARF also target mitochondria for autophagy activation (43, 44). It seems that JNK, like PKR/elF2α (double-stranded RNA-activated protein kinase/eukaryotic initiation factor-2 alpha), positively regulates autophagy by inducing autophagy-related gene expression (–47). Currently, very little is known about how p38 MAPK, ERK/GAIP (extracellular signal-regulated protein kinase/Gα interacting protein), DAPK (death-associated protein kinase) and DRP-1 (death-associated related protein kinase-1) integrate into autophagy (–50). Stimuli to these signaling proteins may alter autophagy activity. However, the final outcome depends not only on these signaling regulators but also on cells and circumstances.
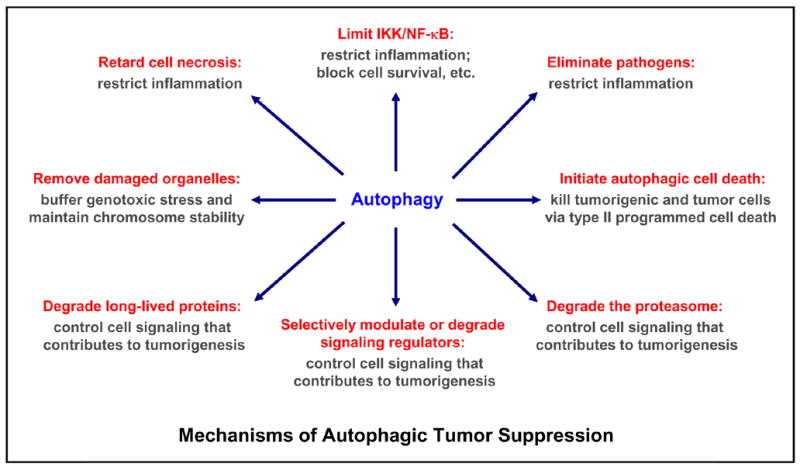
Multiple mechanisms are involved in autophagy-mediated tumor suppression: limit genotoxic damage and maintain chromosome stability by removing damaged or surplus organelles (e.g. mitochondria and endoplasmic reticulum); restrict inflammation by controlling NF-κB activation, blocking cell necrosis, eliminating pathogens; regulating signaling involved in tumorigenesis (particularly, the cell survival and proliferation signaling, e.g. mTOR, NF-κB) by modulating signaling regulatory proteins (e.g. mTOR) or by degrading signaling regulatory proteins (e.g. IKK, NIK), long-lived proteins, proteasome and other organelles; or direct kill harmful cells by type II programmed cell death/autophagic cell death. While autophagy plays an essential role in preventing tumor formation, it may also contribute to tumor progression by allowing tumor cells survival under metabolic stress (low-oxygen and low-nutrient, a common occurrence in tumors). In addition, it may protect some tumor cells against anticancer treatments by blocking the apoptotic pathway and lowering immune-mediated tumor surveillance (also see Figure 5).
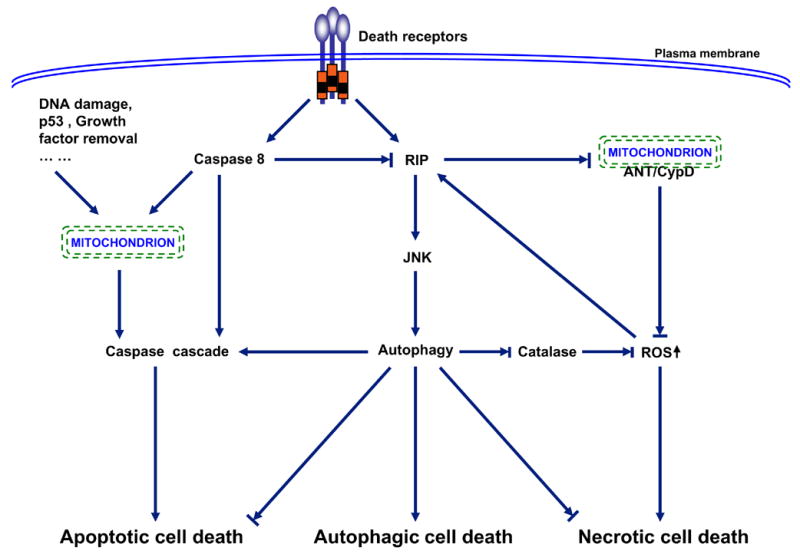
Three major patterns of cell death have been described: apoptosis or type I programmed cell death, autophagy or type II programmed cell death, and necrosis. Hallmarks of apoptotic cell death include activation of caspase cascade, DNA fragmentation, and membrane blebbing. Autophagic cell death is associated with the formation of autophagosomes, double membrane vacuoles that deliver the trapped targets into lysosomes for degradation and recycling. Necrotic cell death is characterized by cellular edema and eventually breakdown of the plasma membrane, leading to release of the cellular components and induction of inflammatory response (90, 91). The three death pathways are interconnected and often entangled. In general, apoptosis activation usually leads to inhibition of both autophagy and necrosis. This inhibition partially attributes to caspase 8-mediated cleavage of RIP1 (receptor interacting protein 1) (–98). One of the important functions of RIP1 in cell autophagy and necrosis is to enhance ROS (reactive oxygen species) production and diminish ATP generation through prevention the interaction between ANT (adenosine nucleotide translocator) and CypD (cyclophilin D) within mitochondrial membrane (–102). Another important function of RIP1 in ROS accumulation is to trigger degradation of catalase, the major enzymatic ROS scavenger, via JNK1-medaited autophagy (55). The activation of autophagy may first start as a survival attempt by cleaning up dysfunctional mitochondira, recycling proteins for energy and nutrient, and blocking necrosis and apoptosis (86, 103). However, when this attempt fails, autophagy may trigger apoptotic cell death, or autophagy itself even becomes cytotoxic, leading to autophagic cell death (–89). This functional shift of autophagy may be due to up-regulation of autophagy-related genes (e.g. Atg4, 5, 7, 12 and beclin 1), leading to over-activation of autophagy (, –105). However, an open mind should be kept that autophagy might only function as a pre-survival mechanism associated with or eventually contributing to cell death; but autophagy itself is not the direct death executor (–89). In tumorigenic and tumor cells, the apoptosis and autophagy are often suppressed. In this case, necrotic cell death becomes dominant. Thus, which cell death ensues really depends on cells and circumstances. In addition, it is also possible that more than one type of cell death may occur simultaneously.
References
-
- Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Exp Biol Med (Maywood) 2006;231:1197–211. - PubMed
-
- Klionsky DJ. Good riddance to bad rubbish. Nature. 2006;441:819–20. - PubMed
-
- Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J. Molecular Cell Biology. 5. 2004. pp. 59–72.pp. 108–30.
-
- Dai C, Whitesell L. HSP90: a rising star on the horizon of anticancer targets. Future Oncol. 2005;1:529–40. - PubMed
-
- Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006;5:596–613. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
