

A recurrent stop-codon mutation in succinate dehydrogenase subunit B gene in normal peripheral blood and childhood T-cell acute leukemia
- PMID: 17487275
- PMCID: PMC1855983
- DOI: 10.1371/journal.pone.0000436
A recurrent stop-codon mutation in succinate dehydrogenase subunit B gene in normal peripheral blood and childhood T-cell acute leukemia
Abstract
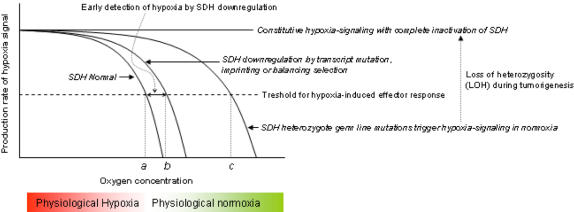
Background: Somatic cytidine mutations in normal mammalian nuclear genes occur during antibody diversification in B lymphocytes and generate an isoform of apolipoprotein B in intestinal cells by RNA editing. Here, I describe that succinate dehydrogenase (SDH; mitochondrial complex II) subunit B gene (SDHB) is somatically mutated at a cytidine residue in normal peripheral blood mononuclear cells (PBMCs) and T-cell acute leukemia. Germ line mutations in the SDHB, SDHC or SDHD genes cause hereditary paraganglioma (PGL) tumors which show constitutive activation of homeostatic mechanisms induced by oxygen deprivation (hypoxia).
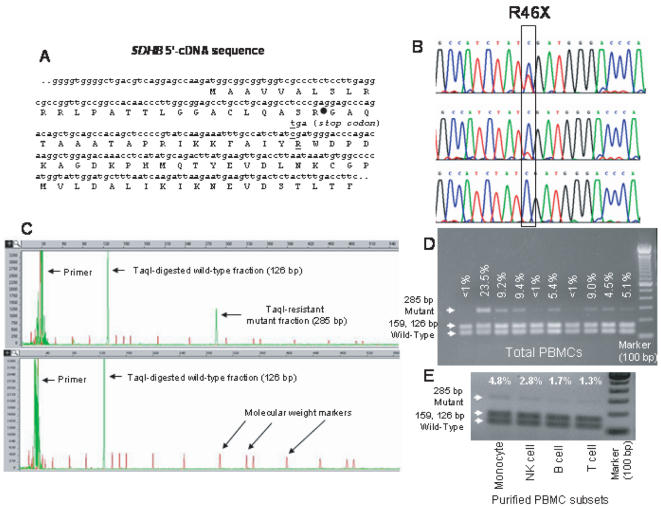
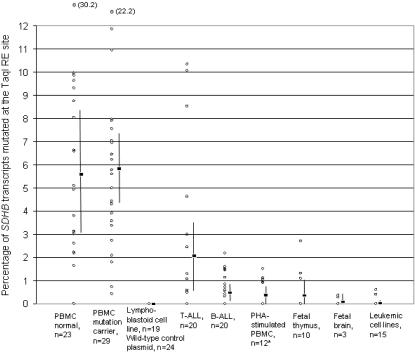
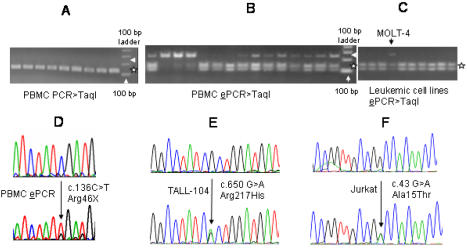
Principal findings: To determine the prevalence of a mutation identified in the SDHB mRNA, 180 samples are tested. An SDHB stop-codon mutation c.136C>T (R46X) is present in a significant fraction (average = 5.8%, range = less than 1 to 30%, n = 52) of the mRNAs obtained from PBMCs. In contrast, the R46X mutation is present in the genomic DNA of PBMCs at very low levels. Examination of the PBMC cell-type subsets identifies monocytes and natural killer (NK) cells as primary sources of the mutant transcript, although lesser contributions also come from B and T lymphocytes. Transcript sequence analyses in leukemic cell lines derived from monocyte, NK, T and B cells indicate that the mutational mechanism targeting SDHB is operational in T-cell acute leukemia. Accordingly, substantial levels (more than 3%) of the mutant SDHB transcripts are detected in five of 20 primary childhood T-cell acute lymphoblastic leukemia (T-ALL) bone marrow samples, but in none of 20 B-ALL samples. In addition, distinct heterozygous SDHB missense DNA mutations are identified in Jurkat and TALL-104 cell lines which are derived from T-ALLs.
Conclusions: The identification of a recurrent, inactivating stop-codon mutation in the SDHB gene in normal blood cells suggests that SDHB is targeted by a cytidine deaminase enzyme. The SDHB mutations in normal PBMCs and leukemic T cells might play a role in cellular pre-adaptation to hypoxia.
Conflict of interest statement
Figures
Similar articles
-
Hypoxia-inducible C-to-U coding RNA editing downregulates SDHB in monocytes.PeerJ. 2013 Sep 10;1:e152. doi: 10.7717/peerj.152. eCollection 2013. PeerJ. 2013. PMID: 24058882 Free PMC article.
-
Mutation analysis of SDHB and SDHC: novel germline mutations in sporadic head and neck paraganglioma and familial paraganglioma and/or pheochromocytoma.BMC Med Genet. 2006 Jan 11;7:1. doi: 10.1186/1471-2350-7-1. BMC Med Genet. 2006. PMID: 16405730 Free PMC article.
-
Mutation analysis of the SDHB and SDHD genes in pheochromocytomas and paragangliomas: identification of a novel nonsense mutation (Q168X) in the SDHB gene.Endocr J. 2010;57(8):745-50. doi: 10.1507/endocrj.k10e-023. Epub 2010 May 25. Endocr J. 2010. PMID: 20505258
-
Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia.Pathology. 2012 Jun;44(4):285-92. doi: 10.1097/PAT.0b013e3283539932. Pathology. 2012. PMID: 22544211 Review.
-
Clinical and molecular progress in hereditary paraganglioma.J Med Genet. 2008 Nov;45(11):689-94. doi: 10.1136/jmg.2008.058560. J Med Genet. 2008. PMID: 18978332 Review.
Cited by
-
SDHA Germline Mutations in SDH-Deficient GISTs: A Current Update.Genes (Basel). 2023 Mar 4;14(3):646. doi: 10.3390/genes14030646. Genes (Basel). 2023. PMID: 36980917 Free PMC article. Review.
-
APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages.Nat Commun. 2015 Apr 21;6:6881. doi: 10.1038/ncomms7881. Nat Commun. 2015. PMID: 25898173 Free PMC article.
-
Revisiting the TCA cycle: signaling to tumor formation.Trends Mol Med. 2011 Nov;17(11):641-9. doi: 10.1016/j.molmed.2011.06.001. Epub 2011 Jul 20. Trends Mol Med. 2011. PMID: 21764377 Free PMC article. Review.
-
TRANSCRIPTIONAL AND PHOSPHO-PROTEOMIC SCREENS REVEAL STEM CELL ACTIVATION OF INSULIN-RESISTANCE AND TRANSFORMATION PATHWAYS FOLLOWING A SINGLE MINIMALLY TOXIC EPISODE OF ROS.Int J Genomics Proteomics. 2011;2(1):34-49. Int J Genomics Proteomics. 2011. PMID: 21743783 Free PMC article.
-
Neck paraganglioma and follicular lymphoma: a case report.J Med Case Rep. 2019 Dec 20;13(1):376. doi: 10.1186/s13256-019-2323-1. J Med Case Rep. 2019. PMID: 31856921 Free PMC article.
References
-
- Chadwick DJ, Goode J, editors. Signaling pathways in acute oxygen sensing; Novartis Foundation symposium. Hoboken: John Wiley; 2006.
-
- Baysal BE, Ferrell RE, Wilett-Brozick JE, Lawrence EC, Myssiorek D, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. - PubMed
-
- Astrom K, Cohen JE, Willett-Brozick JE, Aston CE, Baysal BE. Altitude is a phenotypic modifier in hereditary paraganglioma type 1: evidence for an oxygen-sensing defect. Hum Genet. 2003;113:228–237. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
