

Porcine reproductive and respiratory syndrome virus induces apoptosis through a mitochondria-mediated pathway
- PMID: 17488647
- PMCID: PMC7127477
- DOI: 10.1016/j.virol.2007.04.001
Porcine reproductive and respiratory syndrome virus induces apoptosis through a mitochondria-mediated pathway
Abstract
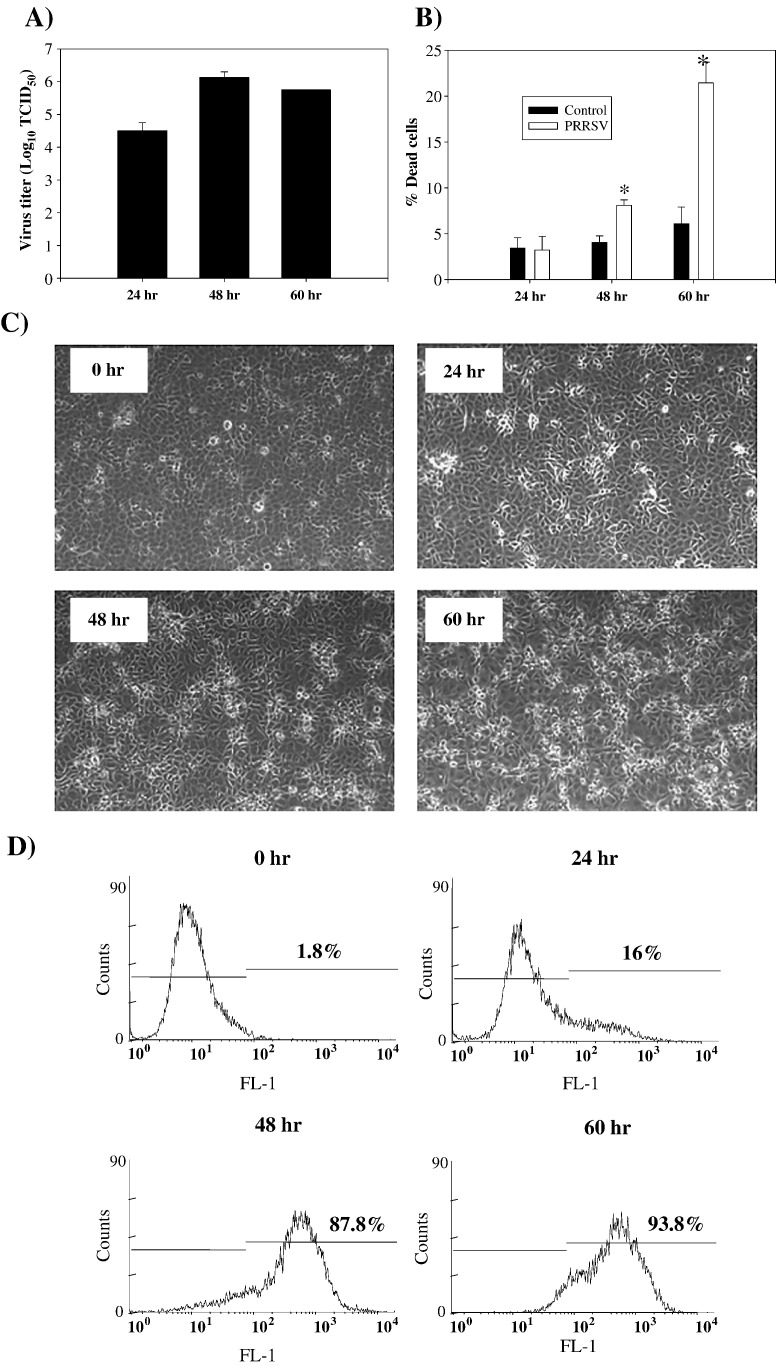
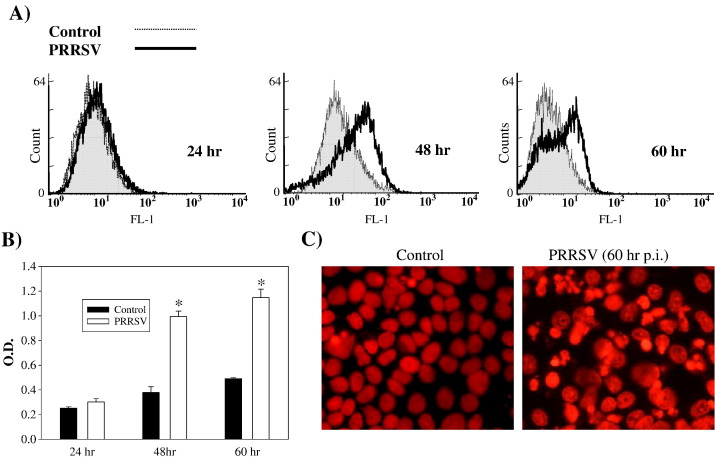
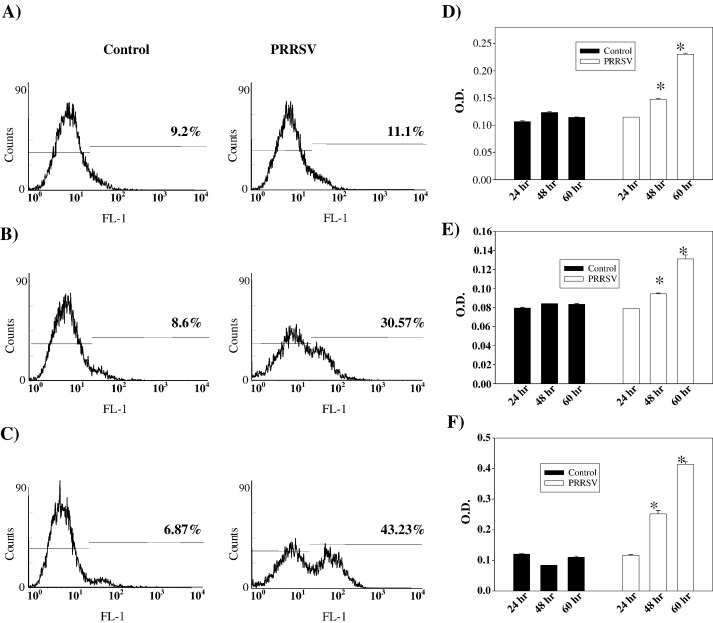
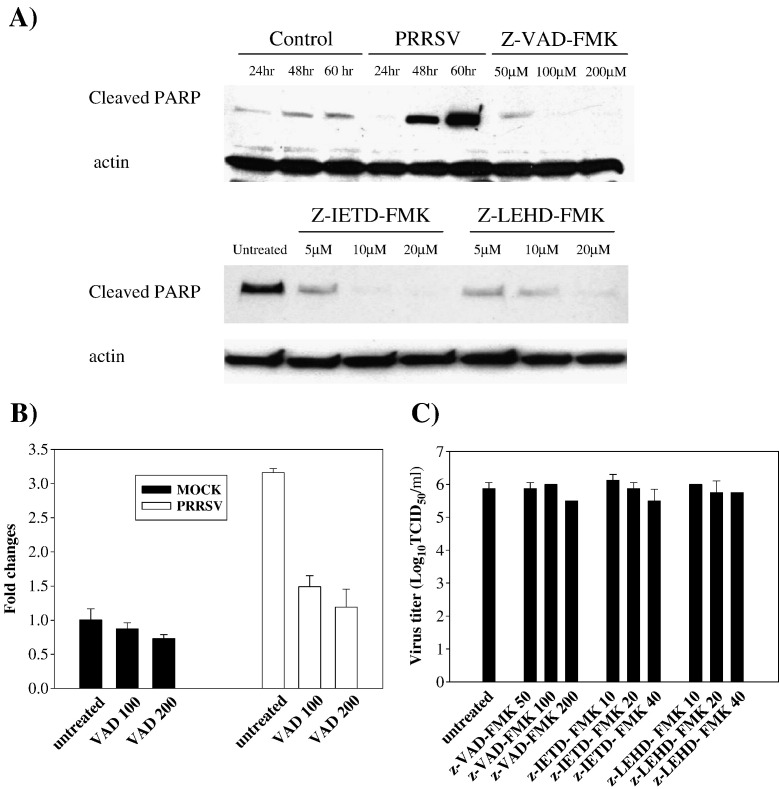
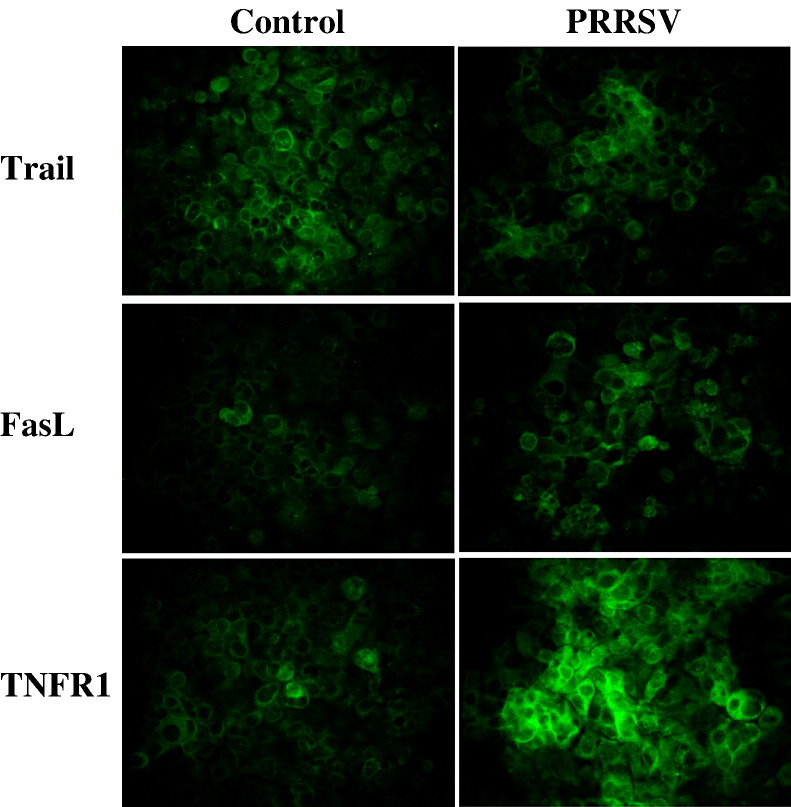
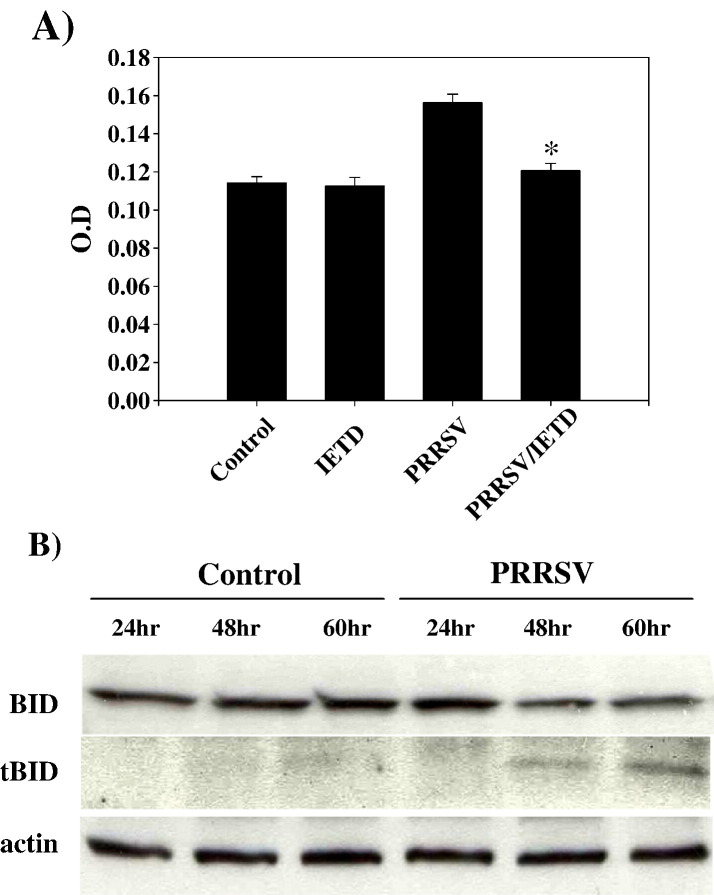
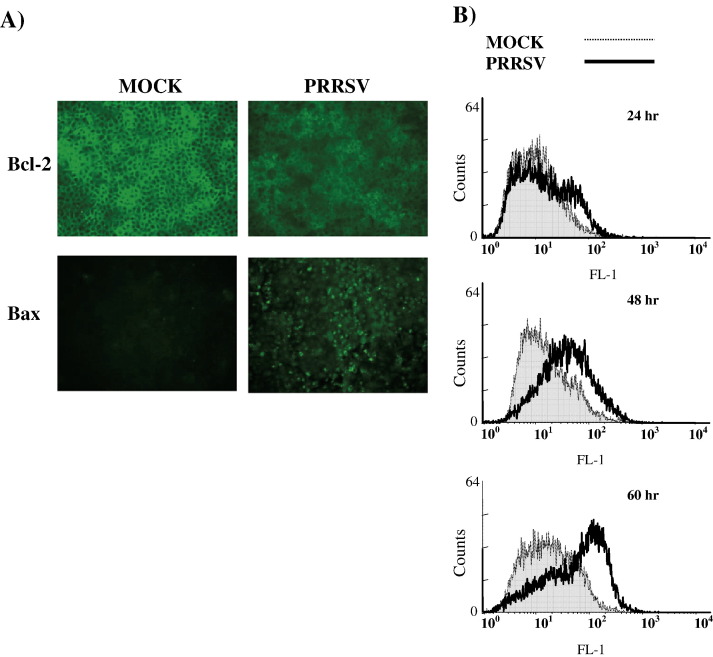
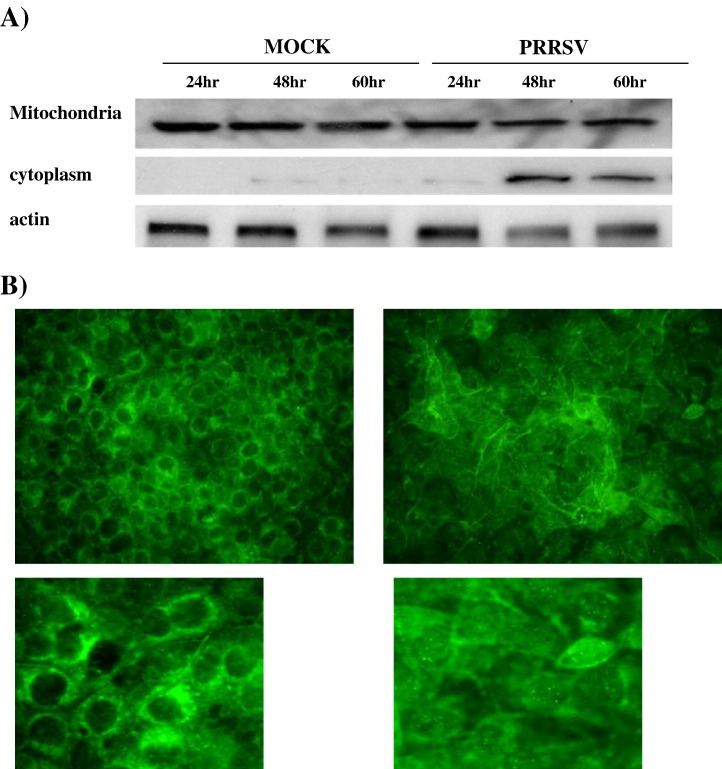
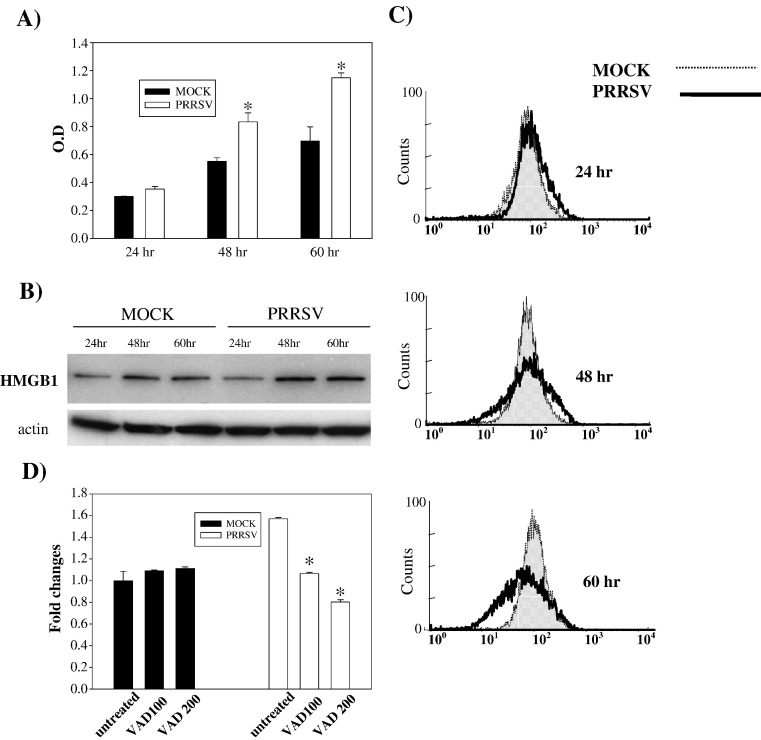
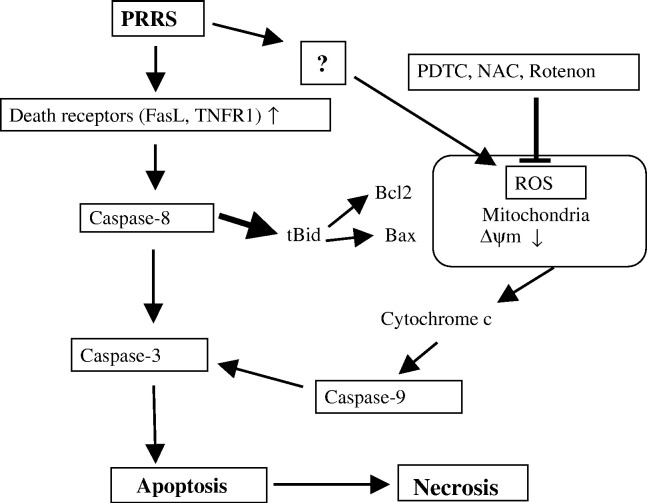
As with a number of other viruses, Porcine reproductive and respiratory syndrome virus (PRRSV) has been shown to induce apoptosis, although the mechanism(s) involved remain unknown. In this study we have characterized the apoptotic pathways activated by PRRSV infection. PRRSV-infected cells showed evidence of apoptosis including phosphatidylserine exposure, chromatin condensation, DNA fragmentation, caspase activation (including caspase-8, 9, 3), and PARP cleavage. DNA fragmentation was dependent on caspase activation but blocking apoptosis by a caspase inhibitor did not affect PRRSV replication. Upregulation of Bax expression by PRRSV infection was followed by disruption of the mitochondria transmembrane potential, resulting in cytochrome c redistridution to the cytoplasm and subsequent caspase-9 activation. A crosstalk between the extrinsic and intrinsic pathways was demonstrated by dependency of caspase-9 activation on active caspase-8 and by Bid cleavage. Furthermore, in this study we provide evidence of the possible involvement of reactive oxygen species (ROS)-mediated oxidative stress in apoptosis induced by PRRSV. Our data indicated that cell death caused by PRRSV infection involves necrosis as well as apoptosis. In summary, these findings demonstrate mechanisms by which PRRSV induces apoptosis and will contribute to an enhanced understanding of PRRSV pathogenesis.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
