

IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system
- PMID: 17492053
- PMCID: PMC1865027
- DOI: 10.1172/JCI30634
IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system
Abstract
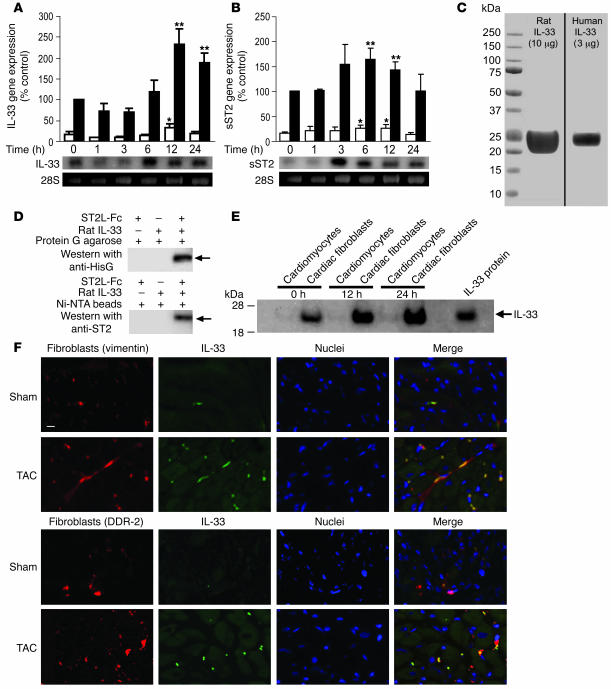
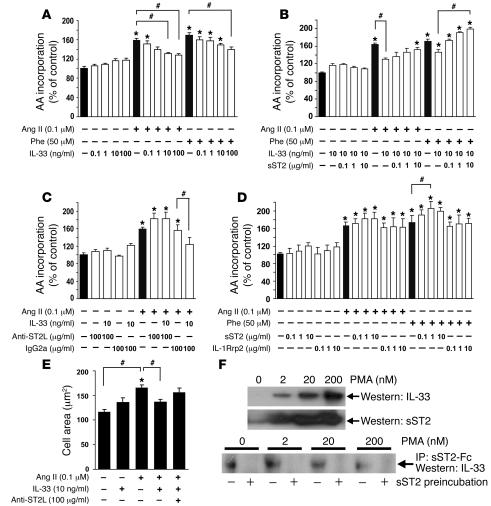
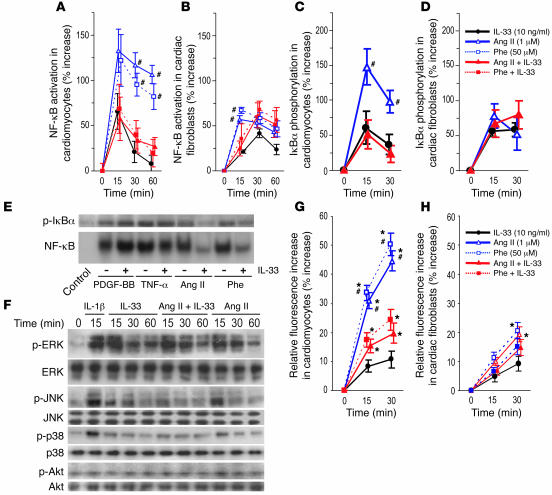
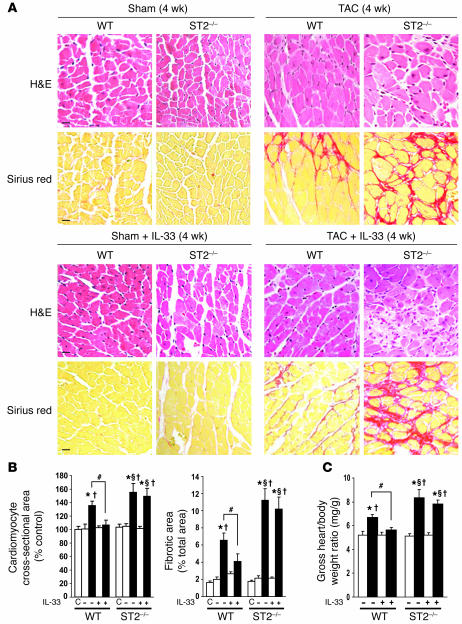
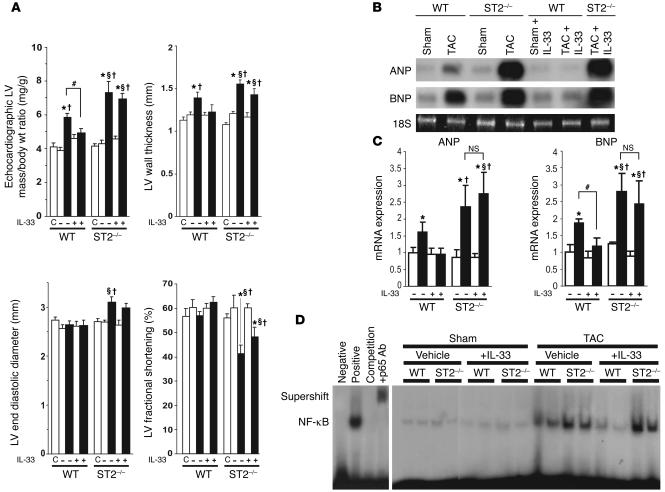
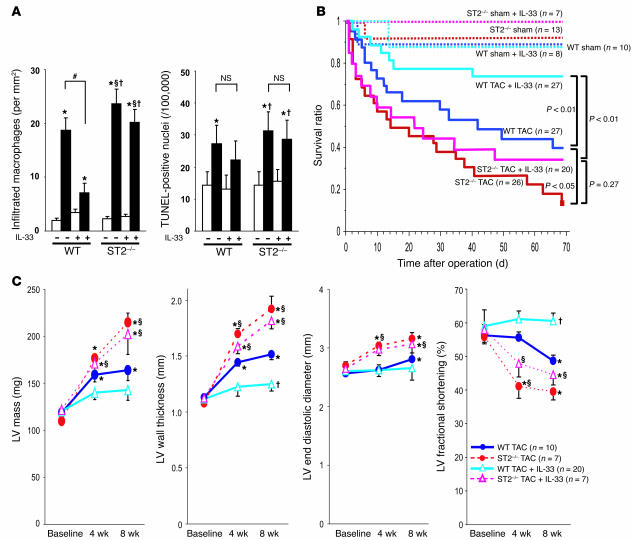
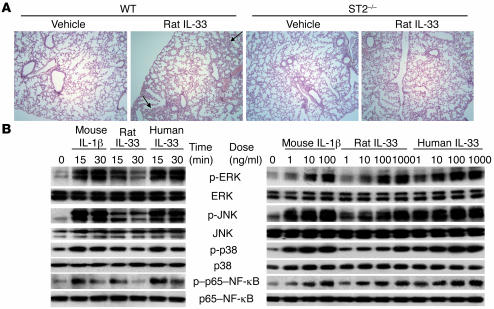
ST2 is an IL-1 receptor family member with transmembrane (ST2L) and soluble (sST2) isoforms. sST2 is a mechanically induced cardiomyocyte protein, and serum sST2 levels predict outcome in patients with acute myocardial infarction or chronic heart failure. Recently, IL-33 was identified as a functional ligand of ST2L, allowing exploration of the role of ST2 in myocardium. We found that IL-33 was a biomechanically induced protein predominantly synthesized by cardiac fibroblasts. IL-33 markedly antagonized angiotensin II- and phenylephrine-induced cardiomyocyte hypertrophy. Although IL-33 activated NF-kappaB, it inhibited angiotensin II- and phenylephrine-induced phosphorylation of inhibitor of NF-kappa B alpha (I kappa B alpha) and NF-kappaB nuclear binding activity. sST2 blocked antihypertrophic effects of IL-33, indicating that sST2 functions in myocardium as a soluble decoy receptor. Following pressure overload by transverse aortic constriction (TAC), ST2(-/-) mice had more left ventricular hypertrophy, more chamber dilation, reduced fractional shortening, more fibrosis, and impaired survival compared with WT littermates. Furthermore, recombinant IL-33 treatment reduced hypertrophy and fibrosis and improved survival after TAC in WT mice, but not in ST2(-/-) littermates. Thus, IL-33/ST2 signaling is a mechanically activated, cardioprotective fibroblast-cardiomyocyte paracrine system, which we believe to be novel. IL-33 may have therapeutic potential for beneficially regulating the myocardial response to overload.
Figures
References
-
- Sadoshima J., Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu. Rev. Physiol. 1997;59:551–571. - PubMed
-
- Diez J., Gonzalez A., Lopez B., Querejeta R. Mechanisms of disease: pathologic structural remodeling is more than adaptive hypertrophy in hypertensive heart disease. Nat. Clin. Pract. Cardiovasc. Med. 2005;2:209–216. - PubMed
-
- Baudino T., Carver W., Giles W.R., Borg T.K. Cardiac fibroblasts: friend or foe? Am. J. Physiol. Heart Circ. Physiol. 2006;291:H1015–H1026. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
