

Contribution of downregulation of L-type calcium currents to delayed neuronal death in rat hippocampus after global cerebral ischemia and reperfusion
- PMID: 17494711
- PMCID: PMC6672382
- DOI: 10.1523/JNEUROSCI.0802-07.2007
Contribution of downregulation of L-type calcium currents to delayed neuronal death in rat hippocampus after global cerebral ischemia and reperfusion
Abstract
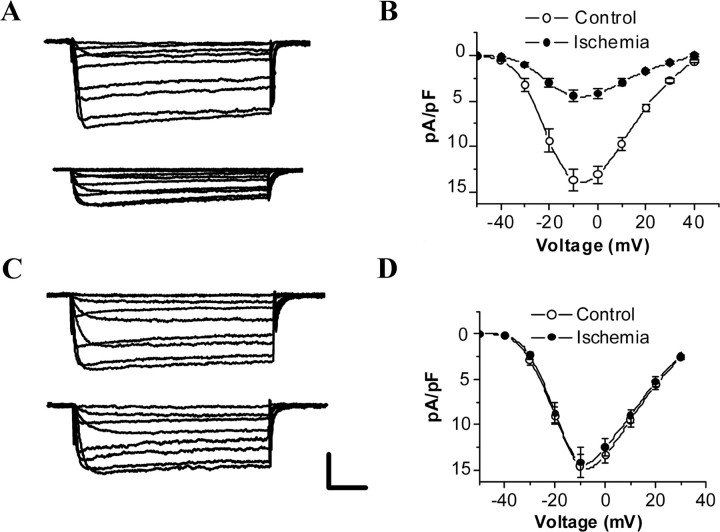
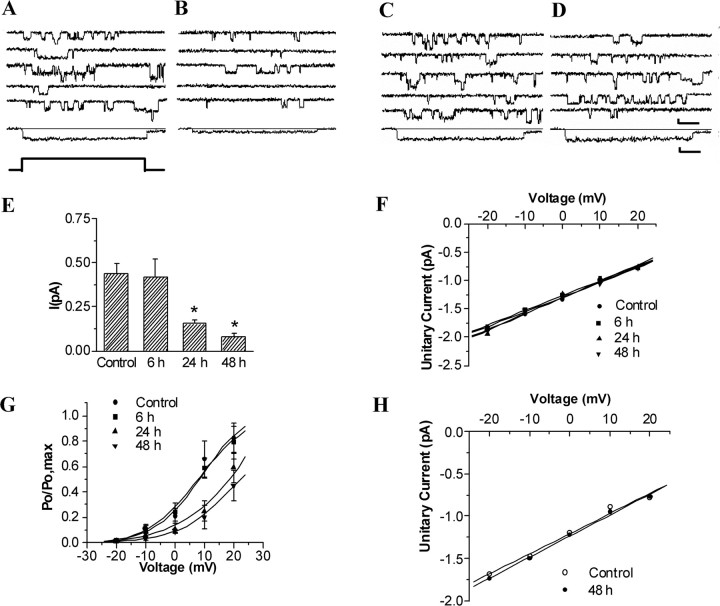
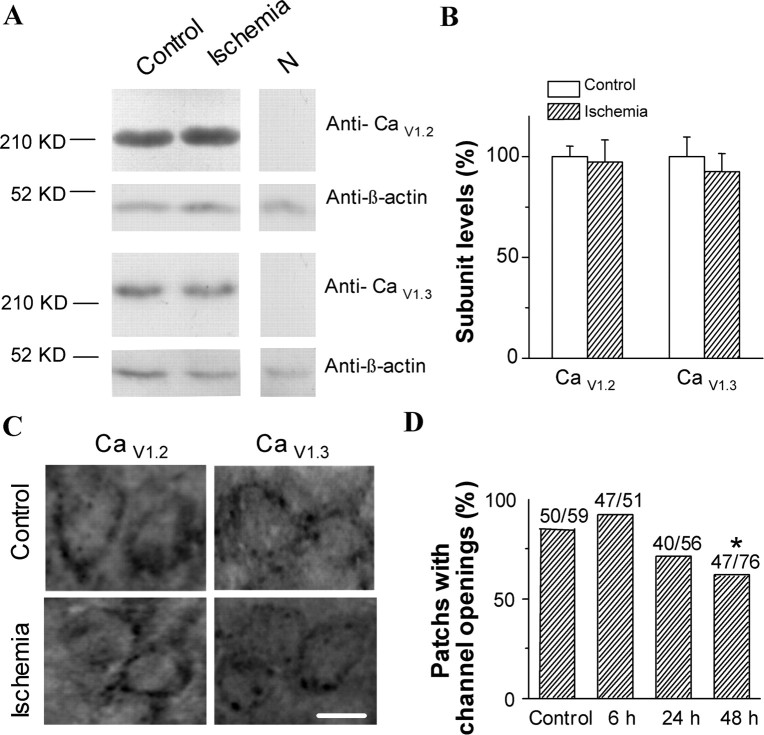
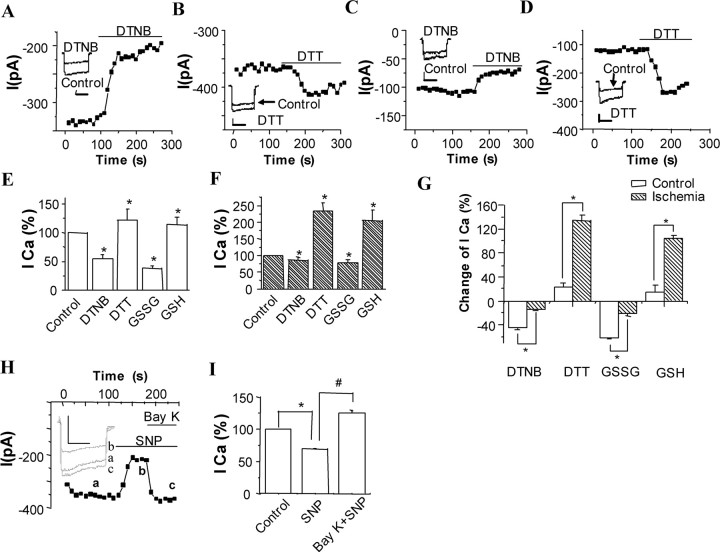
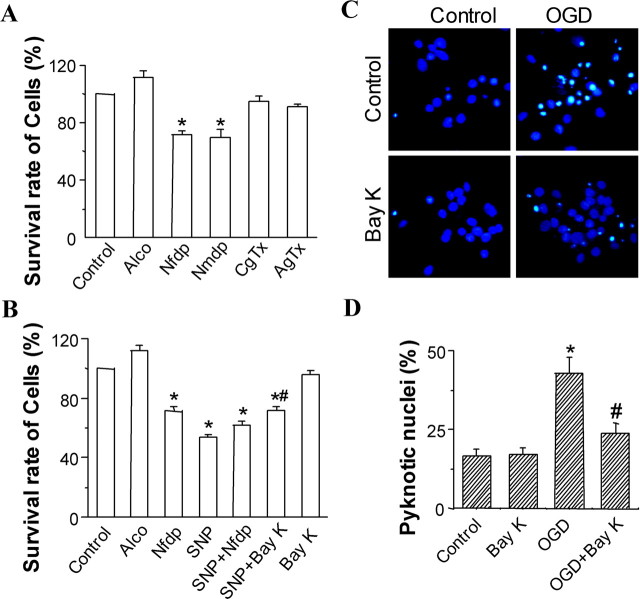
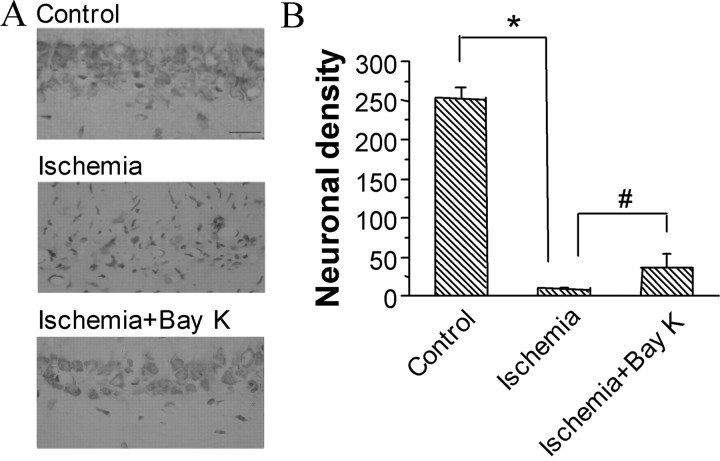
Transient forebrain ischemia induces delayed, selective neuronal death in the CA1 region of the hippocampus. The underlying molecular mechanisms are as yet unclear, but it is known that activation of L-type Ca2+ channels specifically increases the expression of a group of genes required for neuronal survival. Accordingly, we examined temporal changes in L-type calcium-channel activity in CA1 and CA3 pyramidal neurons of rat hippocampus after transient forebrain ischemia by patch-clamp techniques. In vulnerable CA1 neurons, L-type Ca2+-channel activity was persistently downregulated after ischemic insult, whereas in invulnerable CA3 neurons, no change occurred. Downregulation of L-type calcium channels was partially caused by oxidation modulation in postischemic channels. Furthermore, L-type but neither N-type nor P/Q-type Ca2+-channel antagonists alone significantly inhibited the survival of cultured hippocampal neurons. In contrast, specific L-type calcium-channel agonist remarkably reduced neuronal cell death and restored the inhibited channels induced by nitric oxide donor. More importantly, L-type calcium-channel agonist applied after reoxygenation or reperfusion significantly decreased neuronal injury in in vitro oxygen-glucose deprivation ischemic model and in animals subjected to forebrain ischemia-reperfusion. Together, the present results suggest that ischemia-induced inhibition of L-type calcium currents may give rise to delayed death of neurons in the CA1 region, possibly via oxidation mechanisms. Our findings may lead to a new perspective on neuronal death after ischemic insult and suggest that a novel therapeutic approach, activation of L-type calcium channels, could be tested at late stages of reperfusion for stroke treatment.
Figures
Similar articles
-
Transient forebrain ischemia induces persistent hyperactivity of large conductance Ca2+-activated potassium channels via oxidation modulation in rat hippocampal CA1 pyramidal neurons.Eur J Neurosci. 2002 Feb;15(4):779-83. doi: 10.1046/j.1460-9568.2002.01908.x. Eur J Neurosci. 2002. PMID: 11886457
-
Enhancement in activities of large conductance calcium-activated potassium channels in CA1 pyramidal neurons of rat hippocampus after transient forebrain ischemia.Brain Res. 2000 Nov 24;884(1--2):147-54. doi: 10.1016/s0006-8993(00)02923-1. Brain Res. 2000. PMID: 11082496
-
Global ischemia downregulates the function of metabotropic glutamate receptor subtype 5 in hippocampal CA1 pyramidal neurons.Mol Cell Neurosci. 2005 Jul;29(3):484-92. doi: 10.1016/j.mcn.2005.04.001. Mol Cell Neurosci. 2005. PMID: 15882947
-
Neuronal populations of rat cerebral cortex and hippocampus expressed a higher density of L-type Ca 2+ channel than corresponding cerebral vessels.Clin Exp Hypertens. 2002 Oct-Nov;24(7-8):715-26. doi: 10.1081/ceh-120015347. Clin Exp Hypertens. 2002. PMID: 12450246 Review.
-
Pathophysiology and treatment of cerebral ischemia.J Med Invest. 1998 Aug;45(1-4):57-70. J Med Invest. 1998. PMID: 9864965 Review.
Cited by
-
An L-type calcium channel agonist, bay K8644, extends the window of intervention against ischemic neuronal injury.Mol Neurobiol. 2013 Feb;47(1):280-9. doi: 10.1007/s12035-012-8362-x. Epub 2012 Oct 10. Mol Neurobiol. 2013. PMID: 23054684
-
Neuronal injury from cardiac arrest: aging years in minutes.Age (Dordr). 2014;36(4):9680. doi: 10.1007/s11357-014-9680-x. Epub 2014 Aug 8. Age (Dordr). 2014. PMID: 25104136 Free PMC article. Review.
-
Cav3.2 channel regulates cerebral ischemia/reperfusion injury: a promising target for intervention.Neural Regen Res. 2024 Nov 1;19(11):2480-2487. doi: 10.4103/1673-5374.390966. Epub 2023 Dec 15. Neural Regen Res. 2024. PMID: 38526284 Free PMC article.
-
Rhythmic light flicker rescues hippocampal low gamma and protects ischemic neurons by enhancing presynaptic plasticity.Nat Commun. 2020 Jun 15;11(1):3012. doi: 10.1038/s41467-020-16826-0. Nat Commun. 2020. PMID: 32541656 Free PMC article.
-
Gamma frequency entrainment rescues cognitive impairment by decreasing postsynaptic transmission after traumatic brain injury.CNS Neurosci Ther. 2023 Apr;29(4):1142-1153. doi: 10.1111/cns.14096. Epub 2023 Feb 5. CNS Neurosci Ther. 2023. PMID: 36740277 Free PMC article.
References
-
- Andine P, Jacobson I, Hagberg H. Enhanced calcium uptake by CA1 pyramidal cell dendrites in the postischemic phase despite subnormal evoked field potentials: excitatory amino acid receptor dependency and relationship to neuronal damage. J Cereb Blood Flow Metab. 1992;12:773–783. - PubMed
-
- Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;28:275–285. - PubMed
-
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. - PubMed
-
- Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. - PubMed
-
- Burley JR, Dolphin AC. Overlapping selectivity of neurotoxin and dihydropyridine calcium channel blockers in cerebellar granule neurones. Neuropharmacology. 2000;39:1740–1755. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous