

Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation
- PMID: 17494760
- PMCID: PMC1895968
- DOI: 10.1073/pnas.0700420104
Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation
Abstract
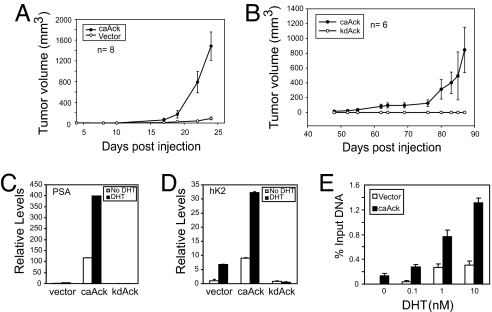
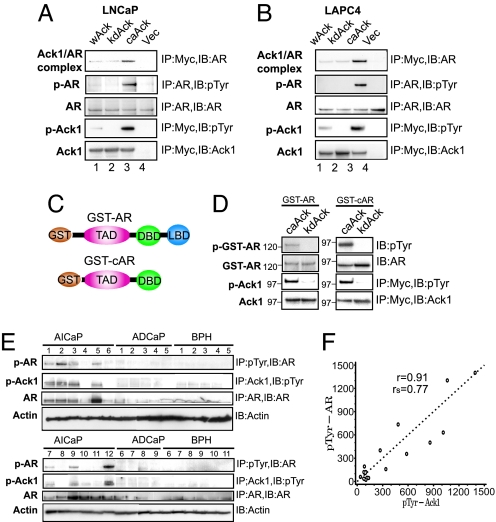
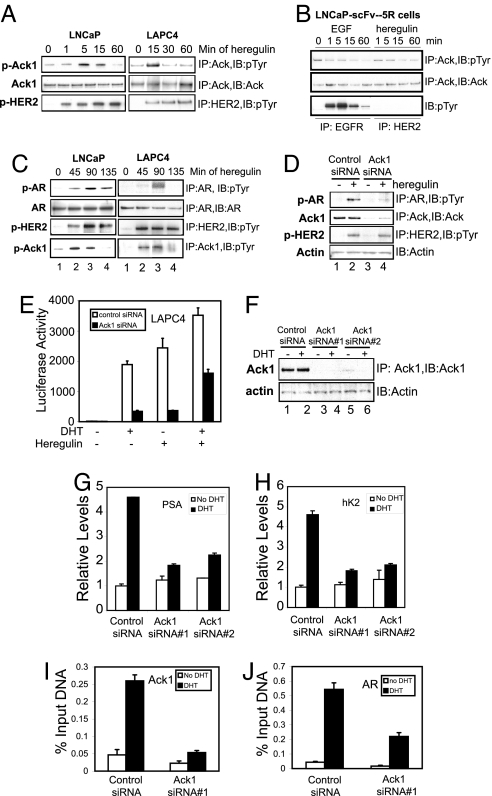
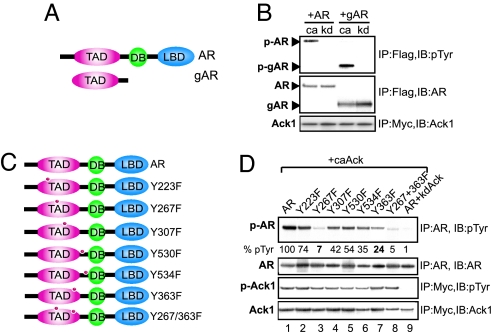
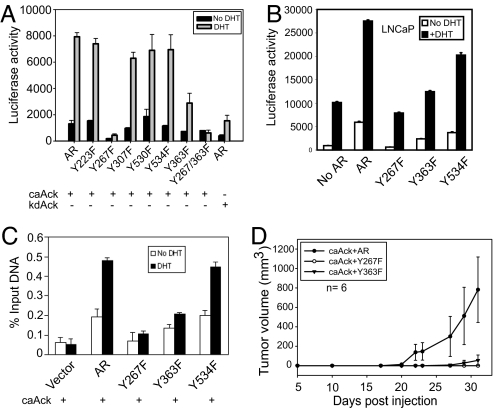
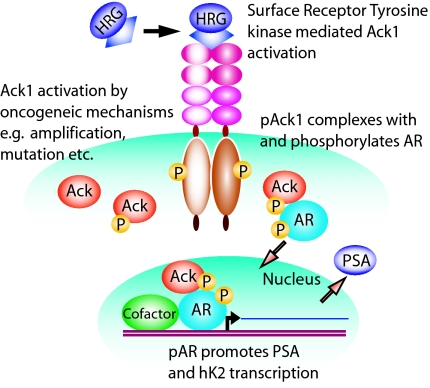
Activation of the androgen receptor (AR) may play a role in androgen-independent progression of prostate cancer. Multiple mechanisms of AR activation, including stimulation by tyrosine kinases, have been postulated. We and others have recently shown involvement of activated Cdc42-associated tyrosine kinase Ack1 in advanced human prostate cancer. Here we provide the molecular basis for interplay between Ack1 and AR in prostate cancer cells. Activated Ack1 promoted androgen-independent growth of LNCaP and LAPC-4 prostate xenograft tumors, AR recruitment to the androgen-responsive enhancer, and androgen-inducible gene expression in the absence of androgen. Heregulin-stimulated HER2 activation induced Ack1 activation and AR tyrosine phosphorylation. Ack1 knockdown inhibited heregulin-dependent AR tyrosine phosphorylation, AR reporter activity, androgen-stimulated gene expression, and AR recruitment. Ack1 was recruited to the androgen-responsive enhancers after androgen and heregulin stimulation. In 8 of 18 primary androgen-independent prostate tumor samples, tyrosine-phosphorylated AR protein was detected and correlated with the detection of tyrosine-phosphorylated Ack1. Neither was elevated in androgen-dependent tumors or benign prostate samples. Activated Ack1 phosphorylated AR protein at Tyr-267 and Tyr-363, both located within the transactivation domain. Mutation of Tyr-267 completely abrogated and mutation of Tyr-363 reduced Ack1-induced AR reporter activation and recruitment of AR to the androgen-responsive enhancer. Expression of AR point mutants inhibited Ack1-driven xenograft tumor growth. Thus, Ack1 activated by surface signals or oncogenic mechanisms may directly enhance AR transcriptional function and promote androgen-independent progression of prostate cancer. Targeting the Ack1 kinase may be a potential therapeutic strategy in prostate cancer.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Effect of Ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity.Prostate. 2010 Sep 1;70(12):1274-85. doi: 10.1002/pros.21163. Prostate. 2010. PMID: 20623637 Free PMC article.
-
Dasatinib inhibits site-specific tyrosine phosphorylation of androgen receptor by Ack1 and Src kinases.Oncogene. 2010 Jun 3;29(22):3208-16. doi: 10.1038/onc.2010.103. Epub 2010 Apr 12. Oncogene. 2010. PMID: 20383201 Free PMC article.
-
Ack1-mediated androgen receptor phosphorylation modulates radiation resistance in castration-resistant prostate cancer.J Biol Chem. 2012 Jun 22;287(26):22112-22. doi: 10.1074/jbc.M112.357384. Epub 2012 May 7. J Biol Chem. 2012. PMID: 22566699 Free PMC article.
-
Shepherding AKT and androgen receptor by Ack1 tyrosine kinase.J Cell Physiol. 2010 Aug;224(2):327-33. doi: 10.1002/jcp.22162. J Cell Physiol. 2010. PMID: 20432460 Free PMC article. Review.
-
ACK1/TNK2 tyrosine kinase: molecular signaling and evolving role in cancers.Oncogene. 2015 Aug 6;34(32):4162-7. doi: 10.1038/onc.2014.350. Epub 2014 Oct 27. Oncogene. 2015. PMID: 25347744 Free PMC article. Review.
Cited by
-
Bioactive natural products for chemoprevention and treatment of castration-resistant prostate cancer.Semin Cancer Biol. 2016 Oct;40-41:160-169. doi: 10.1016/j.semcancer.2016.06.003. Epub 2016 Jun 28. Semin Cancer Biol. 2016. PMID: 27370570 Free PMC article. Review.
-
Effect of Ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity.Prostate. 2010 Sep 1;70(12):1274-85. doi: 10.1002/pros.21163. Prostate. 2010. PMID: 20623637 Free PMC article.
-
Inhibition of Cdc42-intersectin interaction by small molecule ZCL367 impedes cancer cell cycle progression, proliferation, migration, and tumor growth.Cancer Biol Ther. 2019;20(6):740-749. doi: 10.1080/15384047.2018.1564559. Epub 2019 Mar 8. Cancer Biol Ther. 2019. PMID: 30849276 Free PMC article.
-
Minireview: Alternative activation pathways for the androgen receptor in prostate cancer.Mol Endocrinol. 2011 Jun;25(6):897-907. doi: 10.1210/me.2010-0469. Epub 2011 Mar 24. Mol Endocrinol. 2011. PMID: 21436259 Free PMC article. Review.
-
Src controls castration recurrence of CWR22 prostate cancer xenografts.Cancer Med. 2013 Dec;2(6):784-92. doi: 10.1002/cam4.144. Epub 2013 Oct 11. Cancer Med. 2013. PMID: 24403252 Free PMC article.
References
-
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Nat Med. 2004;10:33–39. - PubMed
-
- Grossmann ME, Huang H, Tindall DJ. J Natl Cancer Inst. 2001;93:1687–1697. - PubMed
-
- Dai B, Kim O, Xie Y, Guo Z, Xu K, Wang B, Kong X, Melamed J, Chen H, Bieberich CJ, et al. Cancer Res. 2006;66:8058–8064. - PubMed
-
- Graham DK, Dawson TL, Mullaney DL, Snodgrass HR, Earp HS. Cell Growth Diff. 1994;5:647–657. - PubMed
-
- Mahajan NP, Whang YE, Mohler JL, Earp HS. Cancer Res. 2005;65:10514–10523. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
