

Computational prediction of atomic structures of helical membrane proteins aided by EM maps
- PMID: 17496035
- PMCID: PMC1959528
- DOI: 10.1529/biophysj.106.102137
Computational prediction of atomic structures of helical membrane proteins aided by EM maps
Abstract
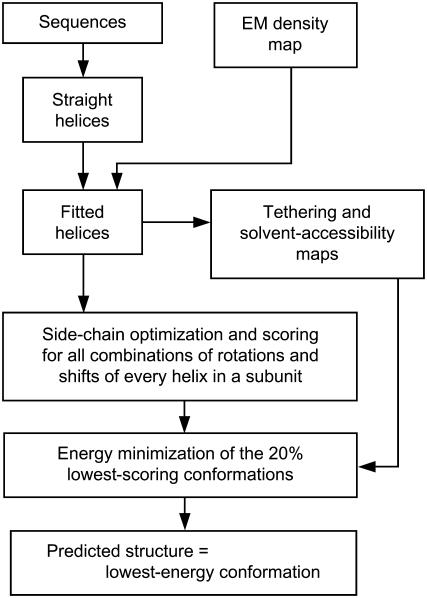
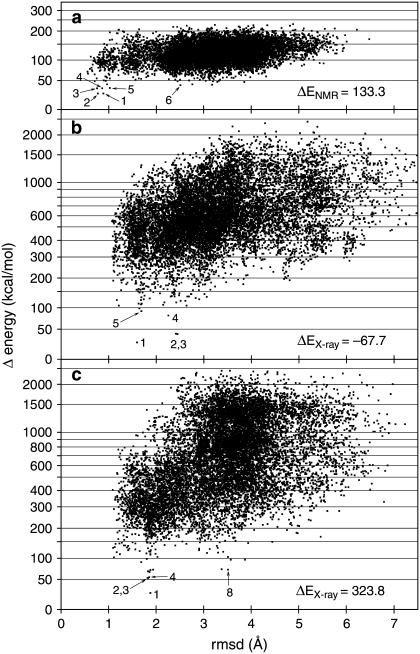
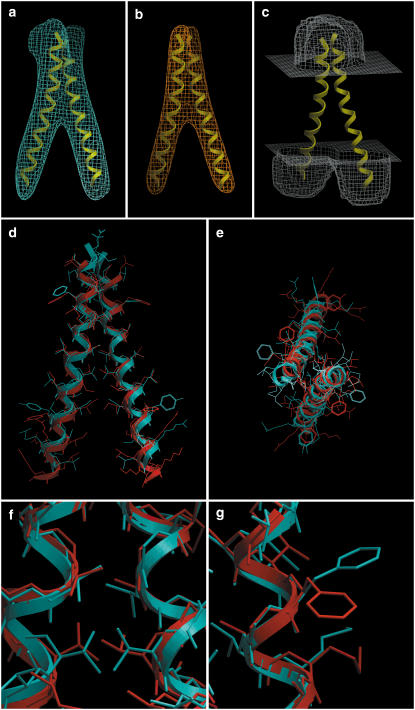
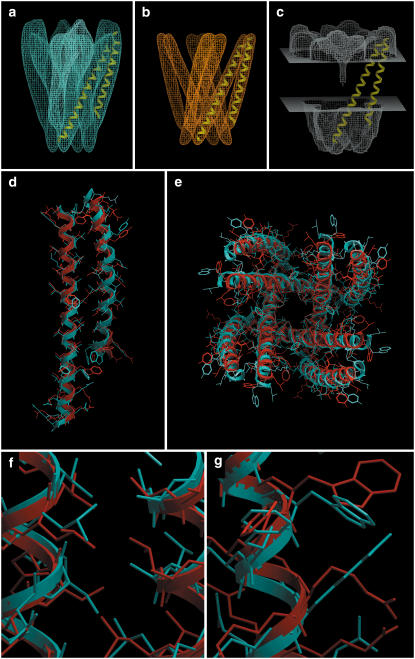
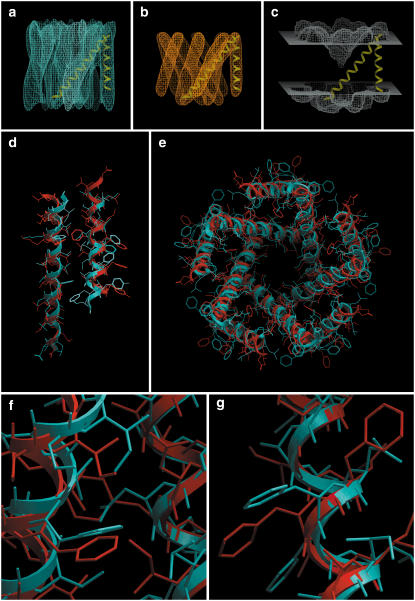
Integral membrane proteins pose a major challenge for protein-structure prediction because only approximately 100 high-resolution structures are available currently, thereby impeding the development of rules or empirical potentials to predict the packing of transmembrane alpha-helices. However, when an intermediate-resolution electron microscopy (EM) map is available, it can be used to provide restraints which, in combination with a suitable computational protocol, make structure prediction feasible. In this work we present such a protocol, which proceeds in three stages: 1), generation of an ensemble of alpha-helices by flexible fitting into each of the density rods in the low-resolution EM map, spanning a range of rotational angles around the main helical axes and translational shifts along the density rods; 2), fast optimization of side chains and scoring of the resulting conformations; and 3), refinement of the lowest-scoring conformations with internal coordinate mechanics, by optimizing the van der Waals, electrostatics, hydrogen bonding, torsional, and solvation energy contributions. In addition, our method implements a penalty term through a so-called tethering map, derived from the EM map, which restrains the positions of the alpha-helices. The protocol was validated on three test cases: GpA, KcsA, and MscL.
Figures
Similar articles
-
Defining the transmembrane helix of M2 protein from influenza A by molecular dynamics simulations in a lipid bilayer.Biophys J. 1999 Apr;76(4):1886-96. doi: 10.1016/s0006-3495(99)77347-9. Biophys J. 1999. PMID: 10096886 Free PMC article.
-
High-resolution crystal structures of protein helices reconciled with three-centered hydrogen bonds and multipole electrostatics.PLoS One. 2015 Apr 20;10(4):e0123146. doi: 10.1371/journal.pone.0123146. eCollection 2015. PLoS One. 2015. PMID: 25894612 Free PMC article.
-
Theoretical and computational models of biological ion channels.Q Rev Biophys. 2004 Feb;37(1):15-103. doi: 10.1017/s0033583504003968. Q Rev Biophys. 2004. PMID: 17390604 Review.
-
Transmembrane alpha-helices in the gap junction membrane channel: systematic search of packing models based on the pair potential function.Microsc Res Tech. 2001 Feb 1;52(3):344-51. doi: 10.1002/1097-0029(20010201)52:3<344::AID-JEMT1018>3.0.CO;2-4. Microsc Res Tech. 2001. PMID: 11180625
-
Emerging issues of connexin channels: biophysics fills the gap.Q Rev Biophys. 2001 Aug;34(3):325-472. doi: 10.1017/s0033583501003705. Q Rev Biophys. 2001. PMID: 11838236 Review.
Cited by
-
Lamin B receptor: multi-tasking at the nuclear envelope.Nucleus. 2010 Jan-Feb;1(1):53-70. doi: 10.4161/nucl.1.1.10515. Nucleus. 2010. PMID: 21327105 Free PMC article. Review.
-
EM-fold: De novo folding of alpha-helical proteins guided by intermediate-resolution electron microscopy density maps.Structure. 2009 Jul 15;17(7):990-1003. doi: 10.1016/j.str.2009.06.001. Structure. 2009. PMID: 19604479 Free PMC article.
-
Blind testing of cross-linking/mass spectrometry hybrid methods in CASP11.Proteins. 2016 Sep;84 Suppl 1(Suppl Suppl 1):152-63. doi: 10.1002/prot.25028. Epub 2016 Mar 28. Proteins. 2016. PMID: 26945814 Free PMC article.
-
Evolutionary bidirectional expansion for the tracing of alpha helices in cryo-electron microscopy reconstructions.J Struct Biol. 2012 Feb;177(2):410-9. doi: 10.1016/j.jsb.2011.11.029. Epub 2011 Dec 6. J Struct Biol. 2012. PMID: 22155667 Free PMC article.
-
Structural refinement of membrane proteins by restrained molecular dynamics and solvent accessibility data.Biophys J. 2008 Dec;95(11):5349-61. doi: 10.1529/biophysj.108.142984. Epub 2008 Aug 1. Biophys J. 2008. PMID: 18676641 Free PMC article.
References
-
- Walker, J. E., and M. Saraste. 1996. Membrane protein structure. Curr. Opin. Struct. Biol. 6:457–459. - PubMed
-
- Drews, J. 2000. Drug discovery: a historical perspective. Science. 287:1960–1964. - PubMed
-
- Bowie, J. U. 2005. Solving the membrane protein folding problem. Nature. 438:581–589. - PubMed
-
- Grisshammer, R. 2006. Understanding recombinant expression of membrane proteins. Curr. Opin. Biotechnol. 17:337–340. - PubMed
-
- Grisshammer, R., and C. Tate. 1995. Overexpression of integral membrane proteins for structural studies. Q. Rev. Biophys. 28:315–422. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
