

Adaptive evolution of a tagged chimeric gammaretrovirus: identification of novel cis-acting elements that modulate splicing
- PMID: 17498744
- PMCID: PMC2938735
- DOI: 10.1016/j.jmb.2007.04.026
Adaptive evolution of a tagged chimeric gammaretrovirus: identification of novel cis-acting elements that modulate splicing
Abstract
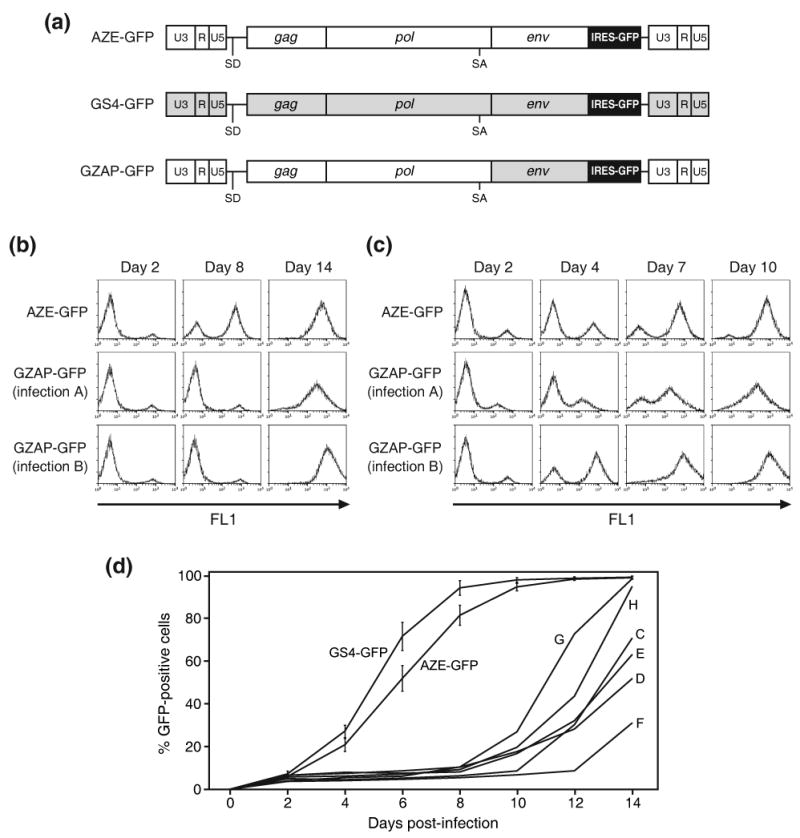
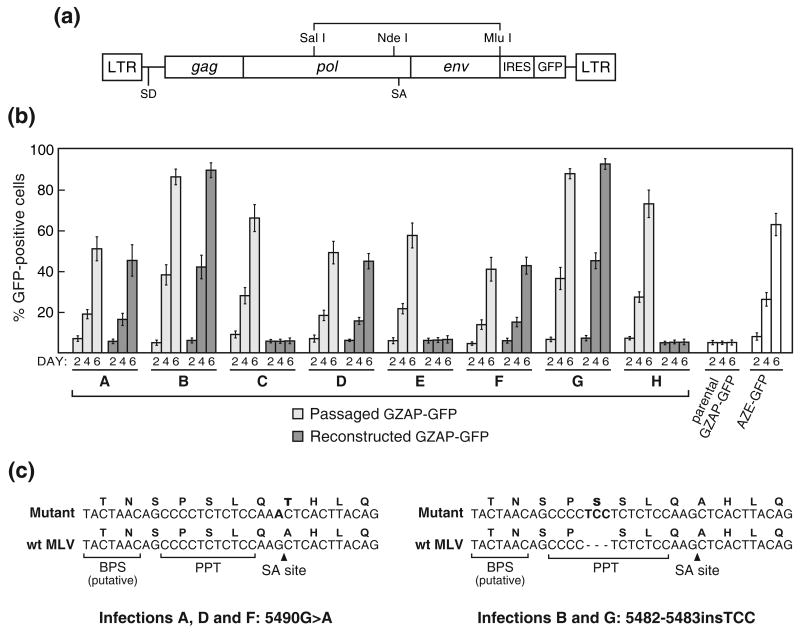
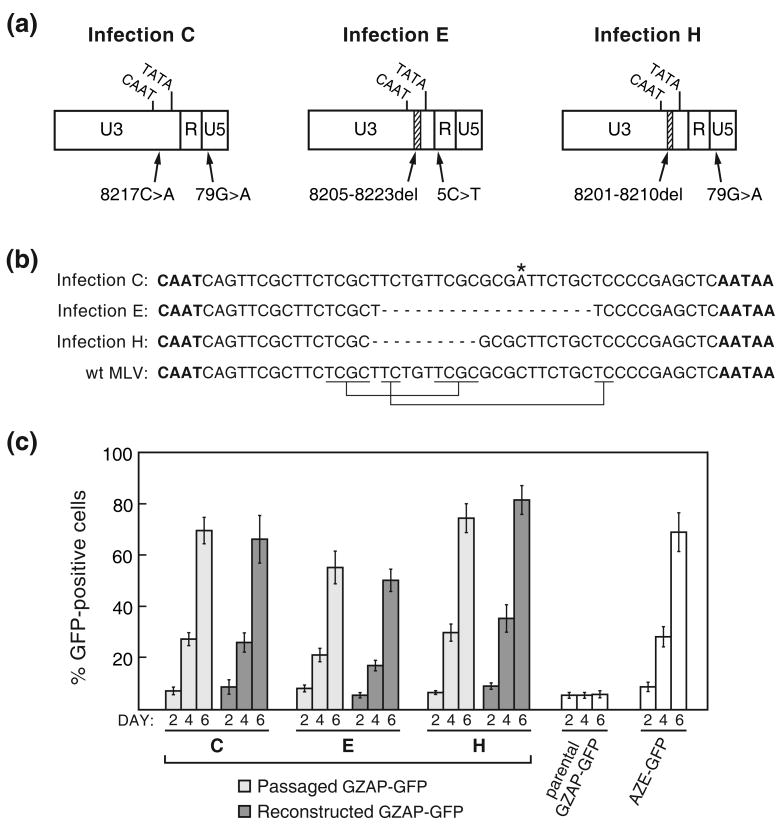
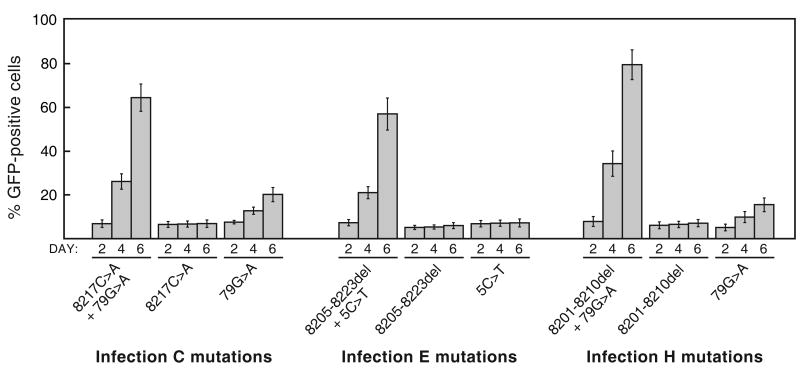
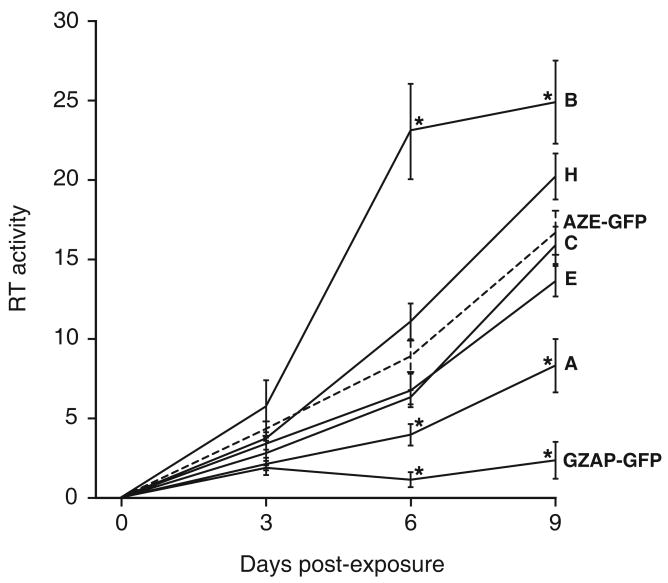
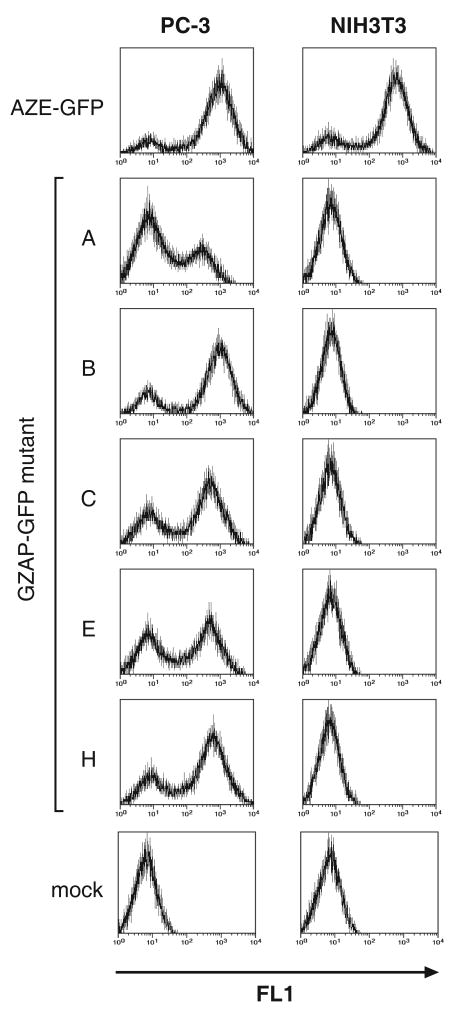
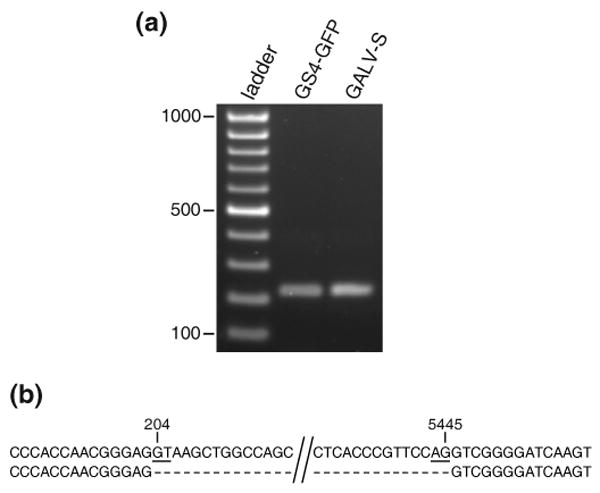
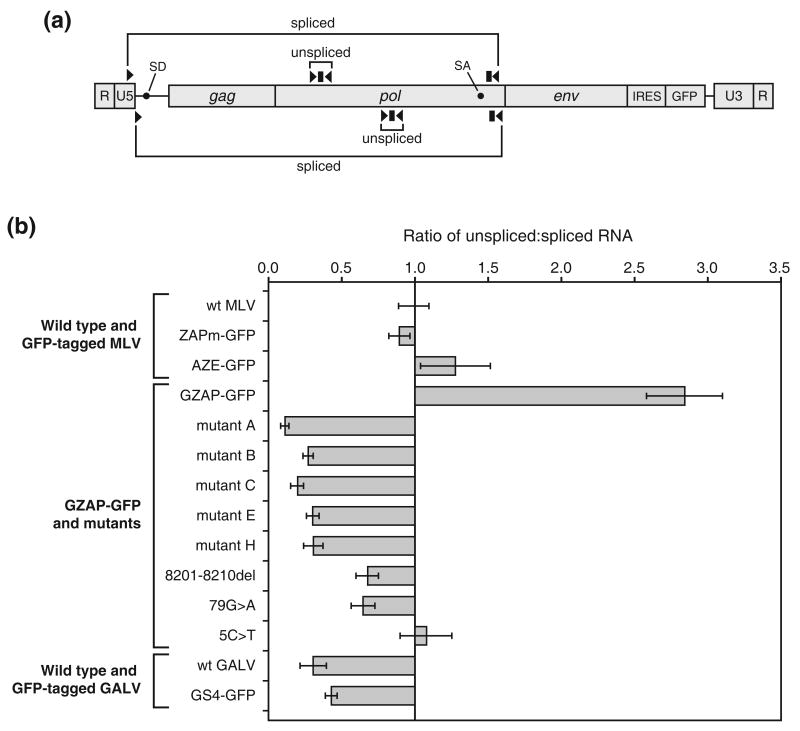
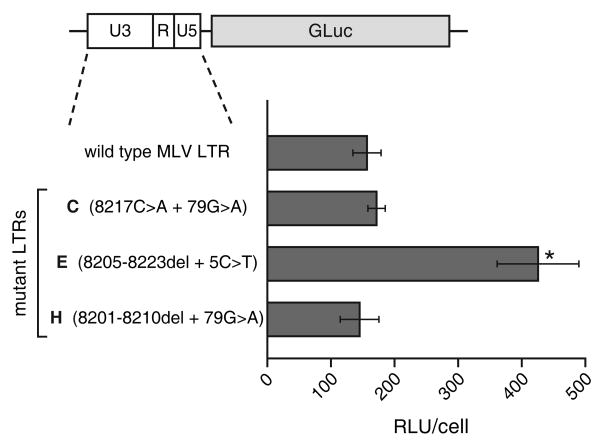
Retroviruses are well known for their ability to incorporate envelope (Env) proteins from other retroviral strains and genera, and even from other virus families. This characteristic has been widely exploited for the generation of replication-defective retroviral vectors, including those derived from murine leukemia virus (MLV), bearing heterologous Env proteins. We investigated the possibility of "genetically pseudotyping" replication-competent MLV by replacing the native env gene in a full-length viral genome with that of another gammaretrovirus. Earlier, we developed replication-competent versions of MLV that stably transmit and express transgenes inserted into the 3' untranslated region of the viral genome. In one such tagged MLV expressing green fluorescent protein, we replaced the native env sequence with that of gibbon ape leukemia virus (GALV). Although the GALV Env protein is commonly used to make high-titer pseudotypes of MLV vectors, we found that the env replacement greatly attenuated viral replication. However, extended cultivation of cells exposed to the chimeric virus resulted in selection of mutants exhibiting rapid replication kinetics and different variants arose in different infections. Two of these variants had acquired mutations at or adjacent to the splice acceptor site, and three others had acquired dual mutations within the long terminal repeat. Analysis of the levels of unspliced and spliced viral RNA produced by the parental and adapted viruses showed that the mutations gained by each of these variants functioned to reverse an imbalance in splicing caused by the env gene substitution. Our results reveal the presence of previously unknown cis-acting sequences in MLV that modulate splicing of the viral transcript and demonstrate that tagging of the retroviral genome with an easily assayed transgene can be combined with in vitro evolution as an approach to efficiently generating and screening for replicating mutants of replication-impaired recombinant viruses.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
