

RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity
- PMID: 17503333
- PMCID: PMC1867103
- DOI: 10.1086/518047
RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity
Erratum in
- Am J Hum Genet. 2007 Nov;81(5):1114. Josifiova, Dragana [corrected to Josifova, Dragana]
Abstract
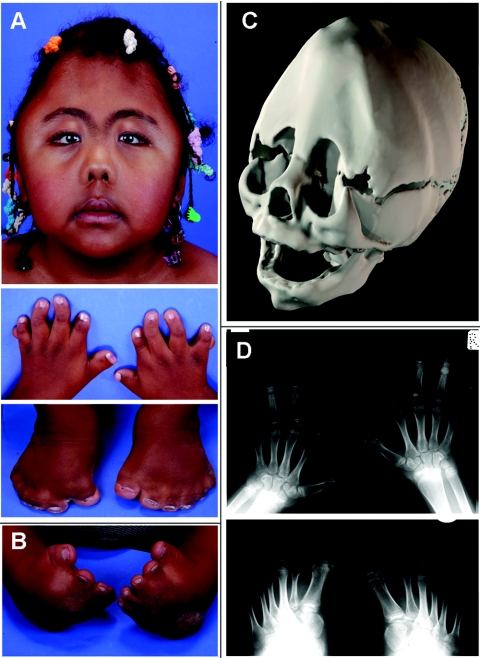
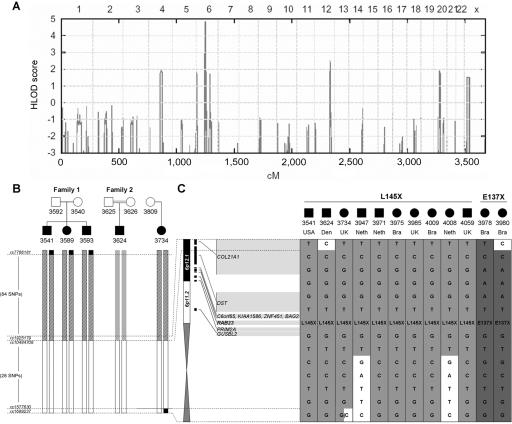
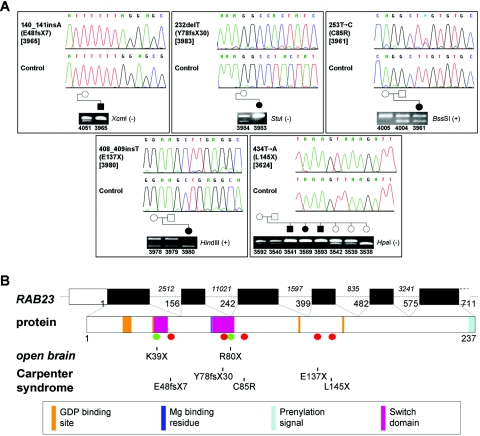
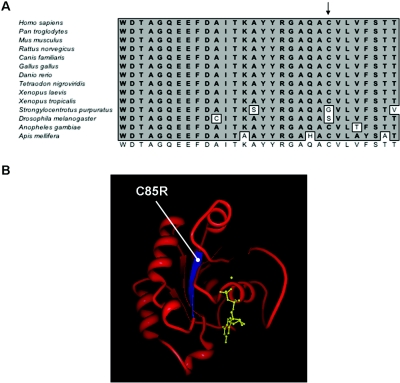
Carpenter syndrome is a pleiotropic disorder with autosomal recessive inheritance, the cardinal features of which include craniosynostosis, polysyndactyly, obesity, and cardiac defects. Using homozygosity mapping, we found linkage to chromosome 6p12.1-q12 and, in 15 independent families, identified five different mutations (four truncating and one missense) in RAB23, which encodes a member of the RAB guanosine triphosphatase (GTPase) family of vesicle transport proteins and acts as a negative regulator of hedgehog (HH) signaling. In 10 patients, the disease was caused by homozygosity for the same nonsense mutation, L145X, that resides on a common haplotype, indicative of a founder effect in patients of northern European descent. Surprisingly, nonsense mutations of Rab23 in open brain mice cause recessive embryonic lethality with neural-tube defects, suggesting a species difference in the requirement for RAB23 during early development. The discovery of RAB23 mutations in patients with Carpenter syndrome implicates HH signaling in cranial-suture biogenesis--an unexpected finding, given that craniosynostosis is not usually associated with mutations of other HH-pathway components--and provides a new molecular target for studies of obesity.
Figures
References
Web Resources
-
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/ (for SNPs, including rs1040461, rs1925179, rs2397214, rs9296842, rs1547625, rs6927258, rs6906792, rs3904827, rs6934928, rs1343391, rs1224703, rs1850417, rs2343013, and rs1689237)
-
- Ensembl Genome Browser, http://www.ensembl.org/ (for RAB23 [reference OTTHUMG00000014918])
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for human RAB23 cDNA reference sequence [accession number NM_183227.1])
-
- International HapMap Project, http://hapmart.hapmap.org/BioMart/martview (for HapMart)
-
- MRC-Holland, http://www.mrc-holland.com/pages/indexpag.html (for information on MLPA reagents and methods)
References
-
- Carpenter G (1901) Two sisters showing malformations of the skull and other congenital abnormalities. Rep Soc Study Dis Child Lond 1:110–118
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
