

PKCepsilon increases phosphorylation of the cardiac myosin binding protein C at serine 302 both in vitro and in vivo
- PMID: 17503784
- PMCID: PMC3969456
- DOI: 10.1021/bi700467k
PKCepsilon increases phosphorylation of the cardiac myosin binding protein C at serine 302 both in vitro and in vivo
Abstract
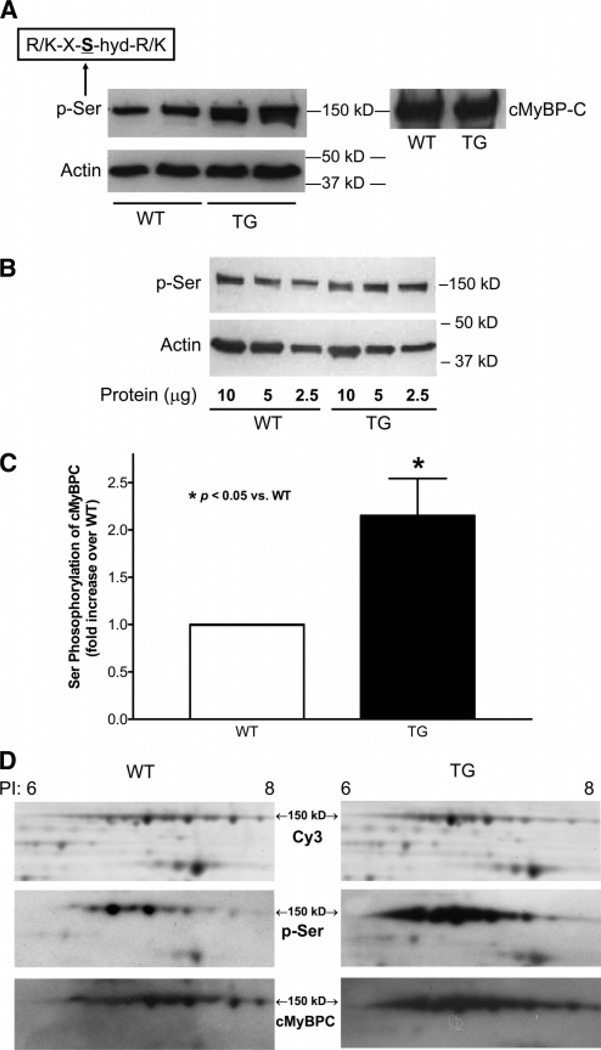
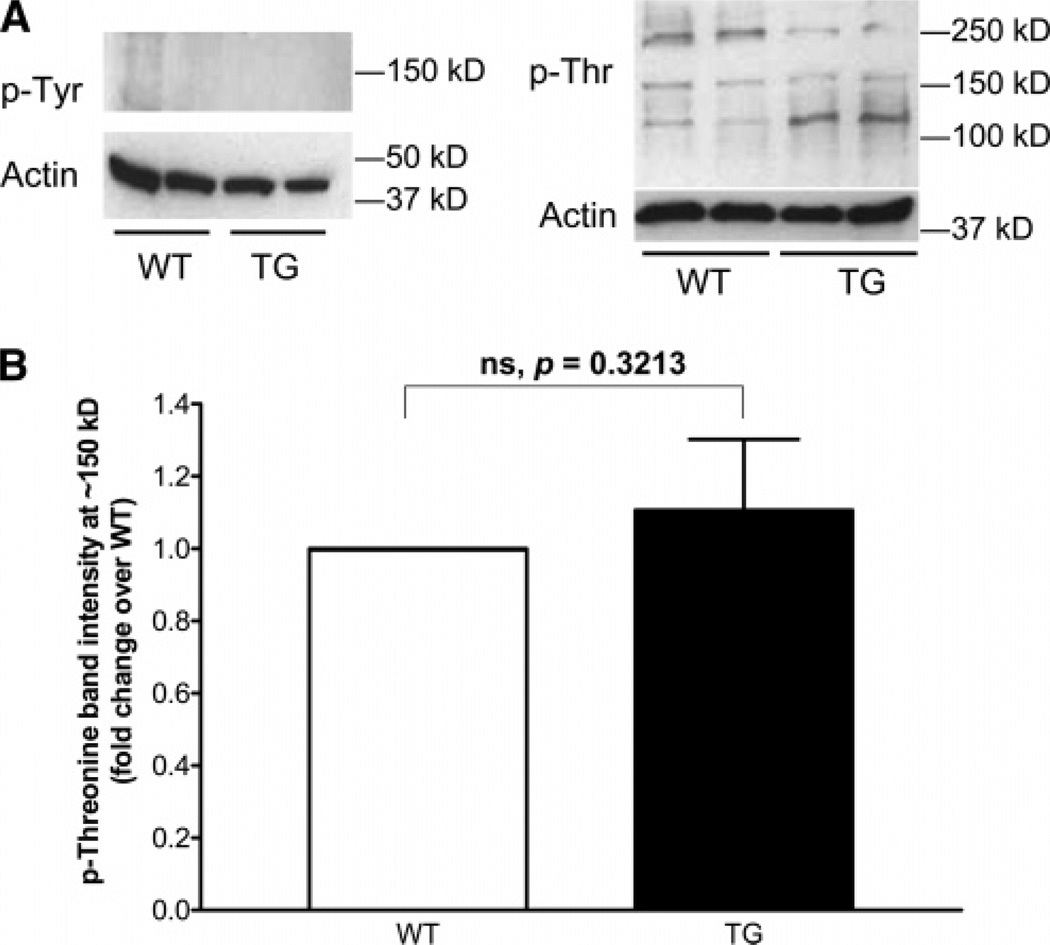
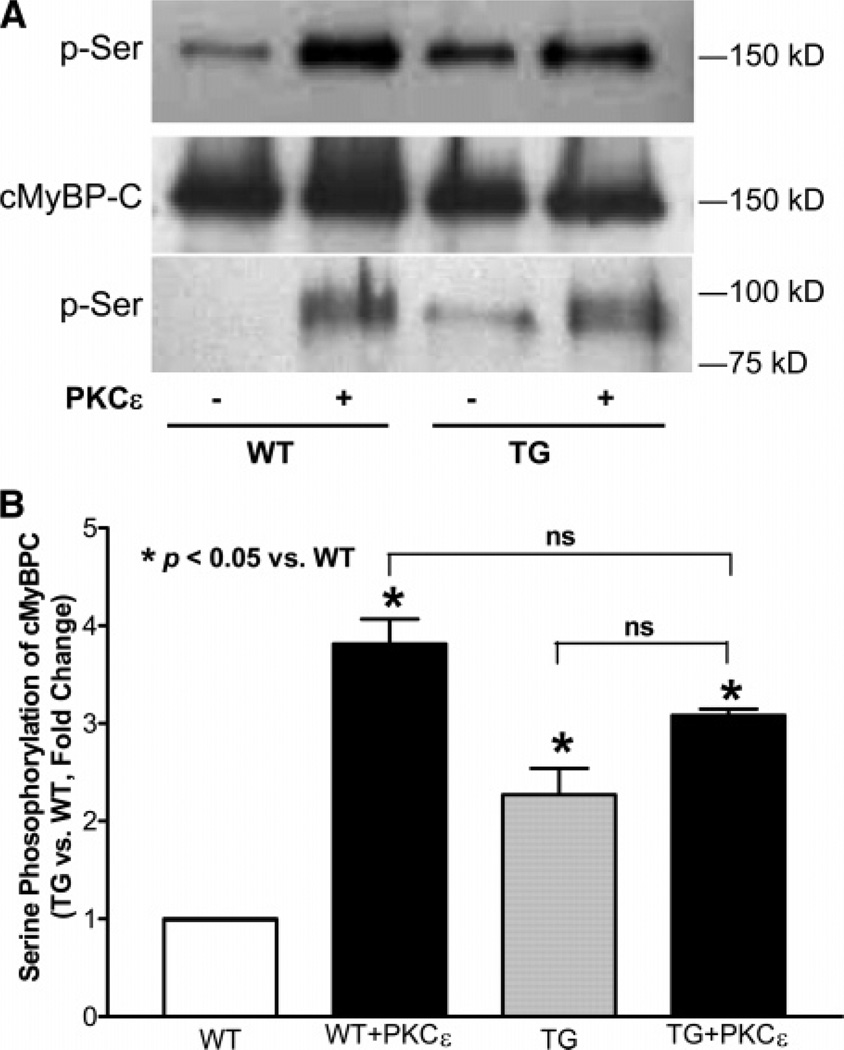
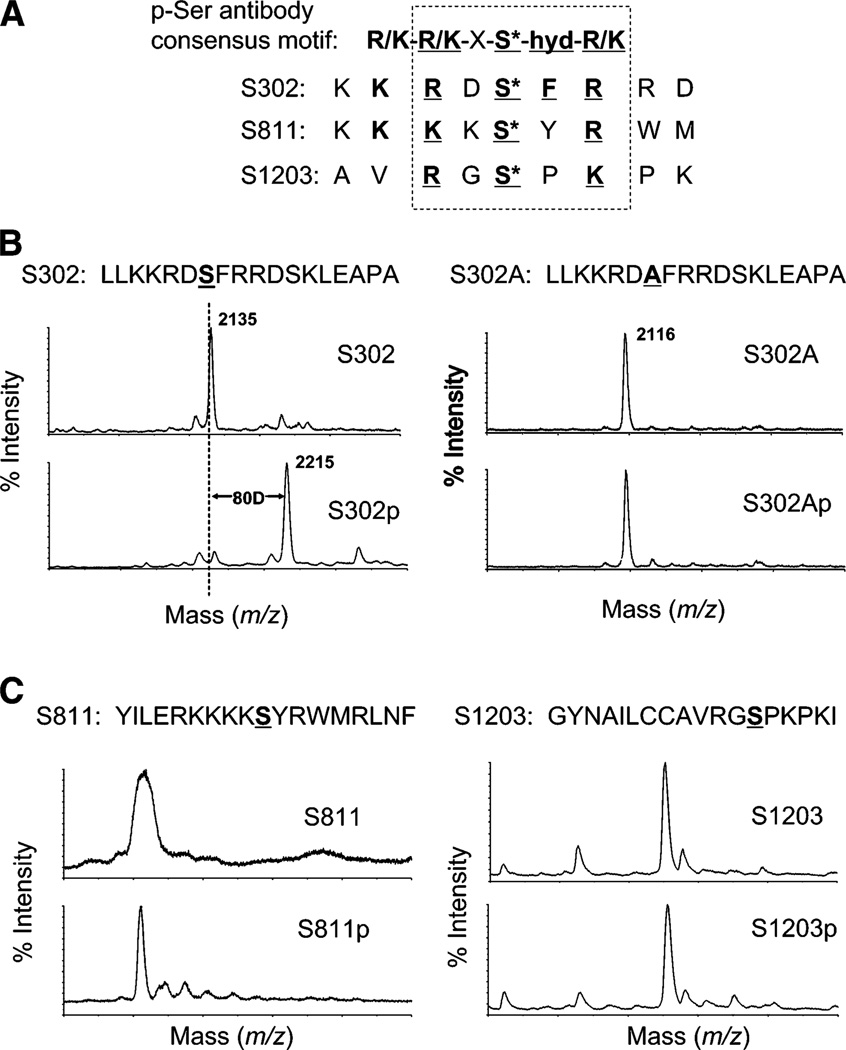
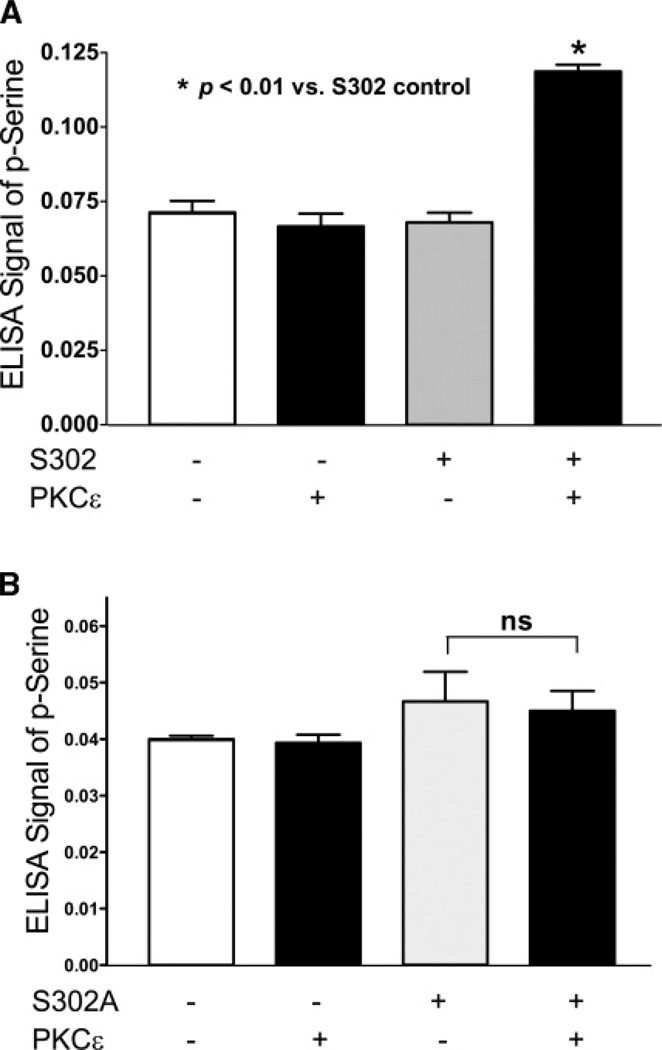
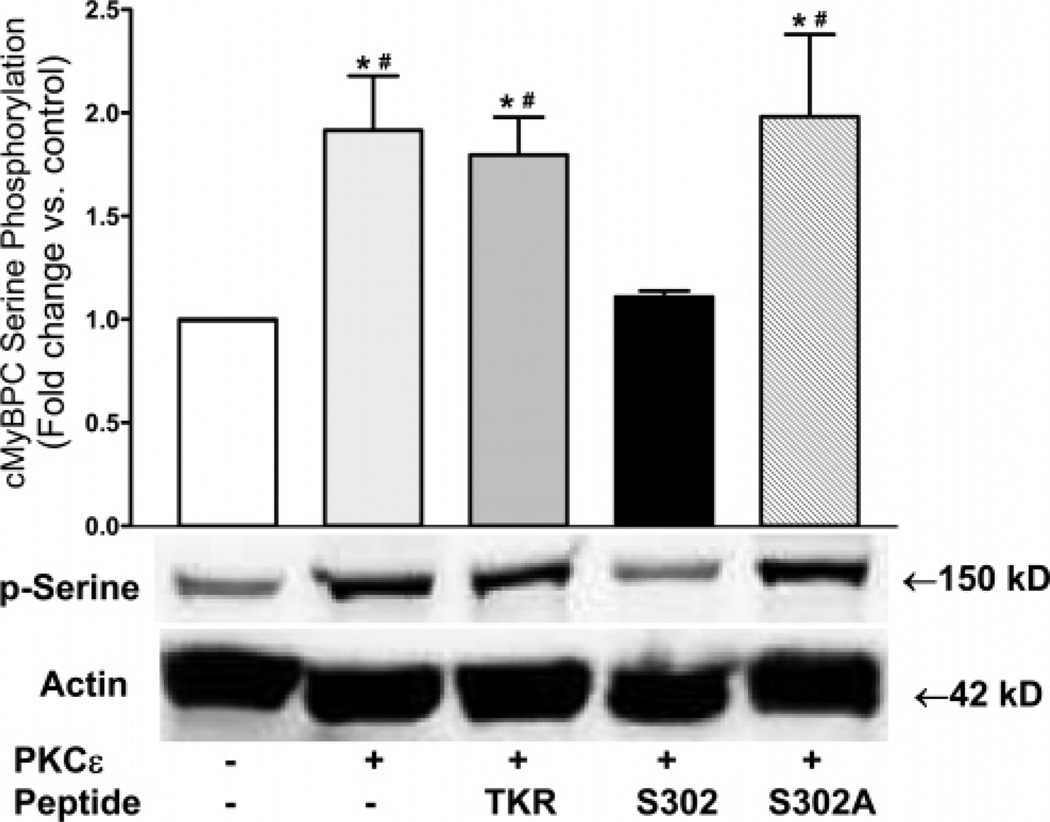
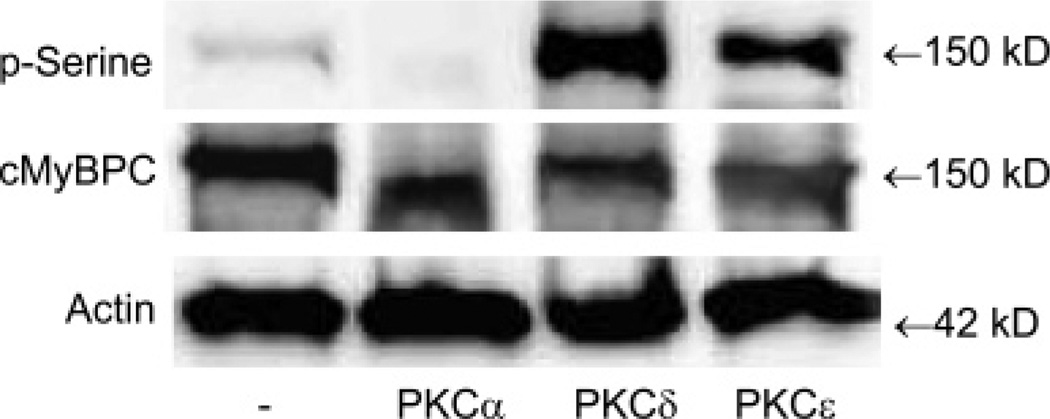
Cardiac myosin binding protein C (cMyBPC) phosphorylation is essential for normal cardiac function. Although PKC was reported to phosphorylate cMyBPC in vitro, the relevant PKC isoforms and functions of PKC-mediated cMyBPC phosphorylation are unknown. We recently reported that a transgenic mouse model with cardiac-specific overexpression of PKCepsilon (PKCepsilon TG) displayed enhanced sarcomeric protein phosphorylation and dilated cardiomyopathy. In the present study, we have investigated cMyBPC phosphorylation in PKCepsilon TG mice. Western blotting and two-dimensional gel electrophoresis demonstrated a significant increase in cMyBPC serine (Ser) phosphorylation in 12-month-old TG mice compared to wild type (WT). In vitro PKCepsilon treatment of myofibrils increased the level of cMyBPC Ser phosphorylation in WT mice to that in TG mice, whereas treatment of TG myofibrils with PKCepsilon showed only a minimal increase in cMyBPC Ser phosphorylation. Three peptide motifs of cMyBPC were identified as the potential PKCepsilon consensus sites including a 100% matched motif at Ser302 and two nearly matched motifs at Ser811 and Ser1203. We treated synthetic peptides corresponding to the sequences of these three motifs with PKCepsilon and determined phosphorylation by mass spectrometry and ELISA assay. PKCepsilon induced phosphorylation at the Ser302 site but not at the Ser811 or Ser1203 sites. A S302A point mutation in the Ser302 peptide abolished the PKCepsilon-dependent phosphorylation. Taken together, our data show that the Ser302 on mouse cMyBPC is a likely PKCepsilon phosphorylation site both in vivo and in vitro and may contribute to the dilated cardiomyopathy associated with increased PKCepsilon activity.
Figures
Similar articles
-
The identification and characterization of novel PKCepsilon phosphorylation sites provide evidence for functional cross-talk within the PKC superfamily.Biochem J. 2008 Apr 15;411(2):319-31. doi: 10.1042/bj20071348. Biochem J. 2008. PMID: 18237277
-
Partial replacement of cardiac troponin I with a non-phosphorylatable mutant at serines 43/45 attenuates the contractile dysfunction associated with PKCepsilon phosphorylation.J Mol Cell Cardiol. 2006 Apr;40(4):465-73. doi: 10.1016/j.yjmcc.2005.12.009. Epub 2006 Jan 30. J Mol Cell Cardiol. 2006. PMID: 16445938
-
Recognition of an intra-chain tandem 14-3-3 binding site within PKCepsilon.EMBO Rep. 2009 Sep;10(9):983-9. doi: 10.1038/embor.2009.150. Epub 2009 Aug 7. EMBO Rep. 2009. PMID: 19662078 Free PMC article.
-
The contribution of cardiac myosin binding protein-c Ser282 phosphorylation to the rate of force generation and in vivo cardiac contractility.J Physiol. 2014 Sep 1;592(17):3747-65. doi: 10.1113/jphysiol.2014.276022. Epub 2014 Jun 20. J Physiol. 2014. PMID: 24951619 Free PMC article.
-
Protein kinase Cepsilon interacts with Stat3 and regulates its activation that is essential for the development of skin cancer.Mol Carcinog. 2007 Aug;46(8):646-53. doi: 10.1002/mc.20356. Mol Carcinog. 2007. PMID: 17583567 Review.
Cited by
-
Stage-specific changes in myofilament protein phosphorylation following myocardial infarction in mice.J Mol Cell Cardiol. 2010 Jun;48(6):1180-6. doi: 10.1016/j.yjmcc.2009.09.010. Epub 2009 Sep 30. J Mol Cell Cardiol. 2010. PMID: 19799909 Free PMC article.
-
Phosphorylation of cardiac myosin binding protein C releases myosin heads from the surface of cardiac thick filaments.Proc Natl Acad Sci U S A. 2017 Feb 21;114(8):E1355-E1364. doi: 10.1073/pnas.1614020114. Epub 2017 Feb 6. Proc Natl Acad Sci U S A. 2017. PMID: 28167762 Free PMC article.
-
Phosphorylation of cardiac Myosin-binding protein-C is a critical mediator of diastolic function.Circ Heart Fail. 2015 May;8(3):582-94. doi: 10.1161/CIRCHEARTFAILURE.114.001550. Epub 2015 Mar 4. Circ Heart Fail. 2015. PMID: 25740839 Free PMC article.
-
Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function.Circ Res. 2008 Oct 24;103(9):974-82. doi: 10.1161/CIRCRESAHA.108.177683. Epub 2008 Sep 18. Circ Res. 2008. PMID: 18802026 Free PMC article.
-
Reduced force production during low blood flow to the heart correlates with altered troponin I phosphorylation.J Muscle Res Cell Motil. 2009;30(3-4):111-23. doi: 10.1007/s10974-009-9180-2. Epub 2009 Jun 9. J Muscle Res Cell Motil. 2009. PMID: 19507043 Free PMC article.
References
-
- Rybin VO, Steinberg SF. Protein kinase C isoform expression and regulation in the developing rat heart. Circ. Res. 1994;74:299–309. - PubMed
-
- Rybin V, Steinberg SF. Do adult rat ventricular myocytes express protein kinase C-alpha? Am. J. Physiol. 1997;272:H2485–H2491. - PubMed
-
- Dorn GW, Robbins J, Sugden PH. Phenotyping hypertrophy: eschew obfuscation. Circ. Res. 2003;92:1171–1175. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
