

A K ATP channel-dependent pathway within alpha cells regulates glucagon release from both rodent and human islets of Langerhans
- PMID: 17503968
- PMCID: PMC1868042
- DOI: 10.1371/journal.pbio.0050143
A K ATP channel-dependent pathway within alpha cells regulates glucagon release from both rodent and human islets of Langerhans
Abstract
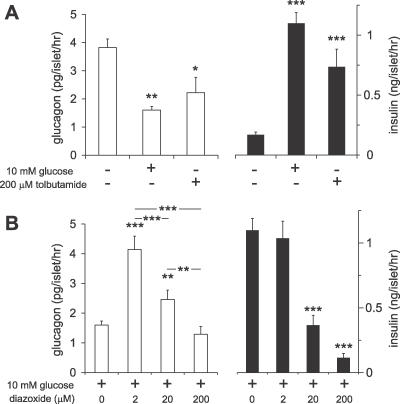
Glucagon, secreted from pancreatic islet alpha cells, stimulates gluconeogenesis and liver glycogen breakdown. The mechanism regulating glucagon release is debated, and variously attributed to neuronal control, paracrine control by neighbouring beta cells, or to an intrinsic glucose sensing by the alpha cells themselves. We examined hormone secretion and Ca(2+) responses of alpha and beta cells within intact rodent and human islets. Glucose-dependent suppression of glucagon release persisted when paracrine GABA or Zn(2+) signalling was blocked, but was reversed by low concentrations (1-20 muM) of the ATP-sensitive K(+) (KATP) channel opener diazoxide, which had no effect on insulin release or beta cell responses. This effect was prevented by the KATP channel blocker tolbutamide (100 muM). Higher diazoxide concentrations (>/=30 muM) decreased glucagon and insulin secretion, and alpha- and beta-cell Ca(2+) responses, in parallel. In the absence of glucose, tolbutamide at low concentrations (<1 muM) stimulated glucagon secretion, whereas high concentrations (>10 muM) were inhibitory. In the presence of a maximally inhibitory concentration of tolbutamide (0.5 mM), glucose had no additional suppressive effect. Downstream of the KATP channel, inhibition of voltage-gated Na(+) (TTX) and N-type Ca(2+) channels (omega-conotoxin), but not L-type Ca(2+) channels (nifedipine), prevented glucagon secretion. Both the N-type Ca(2+) channels and alpha-cell exocytosis were inactivated at depolarised membrane potentials. Rodent and human glucagon secretion is regulated by an alpha-cell KATP channel-dependent mechanism. We propose that elevated glucose reduces electrical activity and exocytosis via depolarisation-induced inactivation of ion channels involved in action potential firing and secretion.
Conflict of interest statement
Figures









Similar articles
-
Glucose inhibits glucagon secretion by decreasing [Ca2+]c and by reducing the efficacy of Ca2+ on exocytosis via somatostatin-dependent and independent mechanisms.Mol Metab. 2022 Jul;61:101495. doi: 10.1016/j.molmet.2022.101495. Epub 2022 Apr 11. Mol Metab. 2022. PMID: 35421610 Free PMC article.
-
Glucose stimulates glucagon release in single rat alpha-cells by mechanisms that mirror the stimulus-secretion coupling in beta-cells.Endocrinology. 2005 Nov;146(11):4861-70. doi: 10.1210/en.2005-0800. Epub 2005 Aug 4. Endocrinology. 2005. PMID: 16081632
-
KATP channel blockers control glucagon secretion by distinct mechanisms: A direct stimulation of α-cells involving a [Ca2+]c rise and an indirect inhibition mediated by somatostatin.Mol Metab. 2021 Nov;53:101268. doi: 10.1016/j.molmet.2021.101268. Epub 2021 Jun 9. Mol Metab. 2021. PMID: 34118477 Free PMC article.
-
'Resistance is futile?' - paradoxical inhibitory effects of KATP channel closure in glucagon-secreting α-cells.J Physiol. 2020 Nov;598(21):4765-4780. doi: 10.1113/JP279775. Epub 2020 Aug 7. J Physiol. 2020. PMID: 32716554 Free PMC article. Review.
-
Regulation of glucagon secretion by glucose: paracrine, intrinsic or both?Diabetes Obes Metab. 2011 Oct;13 Suppl 1:95-105. doi: 10.1111/j.1463-1326.2011.01450.x. Diabetes Obes Metab. 2011. PMID: 21824262 Review.
Cited by
-
A computational systems analysis of factors regulating α cell glucagon secretion.Islets. 2012 Jul-Aug;4(4):262-83. doi: 10.4161/isl.22193. Epub 2012 Jul 1. Islets. 2012. PMID: 23093806 Free PMC article.
-
Model for glucagon secretion by pancreatic α-cells.PLoS One. 2012;7(3):e32282. doi: 10.1371/journal.pone.0032282. Epub 2012 Mar 7. PLoS One. 2012. PMID: 22412861 Free PMC article.
-
Glucose-mediated inhibition of calcium-activated potassium channels limits α-cell calcium influx and glucagon secretion.Am J Physiol Endocrinol Metab. 2019 Apr 1;316(4):E646-E659. doi: 10.1152/ajpendo.00342.2018. Epub 2019 Jan 29. Am J Physiol Endocrinol Metab. 2019. PMID: 30694690 Free PMC article.
-
Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas.Diabetologia. 2008 Dec;51(12):2263-70. doi: 10.1007/s00125-008-1149-y. Epub 2008 Sep 16. Diabetologia. 2008. PMID: 18795252
-
Modeling the pancreatic α-cell: dual mechanisms of glucose suppression of glucagon secretion.Biophys J. 2014 Feb 4;106(3):741-51. doi: 10.1016/j.bpj.2013.11.4504. Biophys J. 2014. PMID: 24507615 Free PMC article.
References
-
- Shah P, Basu A, Basu R, Rizza R. Impact of lack of suppression of glucagon on glucose tolerance in humans. Am J Physiol. 1999;277:E283–E290. - PubMed
-
- Cryer PE. Hypoglycaemia: The limiting factor in the glycaemic management of Type I and Type II diabetes. Diabetologia. 2002;45:937–948. - PubMed
-
- Cryer PE. Diverse causes of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med. 2004;350:2272–2279. - PubMed
-
- Gromada J, Franklin I, Wollheim CB. α-Cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev. 2007;28:84–116. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
