

The Deuterator: software for the determination of backbone amide deuterium levels from H/D exchange MS data
- PMID: 17506883
- PMCID: PMC1876250
- DOI: 10.1186/1471-2105-8-156
The Deuterator: software for the determination of backbone amide deuterium levels from H/D exchange MS data
Abstract
Background: The combination of mass spectrometry and solution phase amide hydrogen/deuterium exchange (H/D exchange) experiments is an effective method for characterizing protein dynamics, and protein-protein or protein-ligand interactions. Despite methodological advancements and improvements in instrumentation and automation, data analysis and display remains a tedious process. The factors that contribute to this bottleneck are the large number of data points produced in a typical experiment, each requiring manual curation and validation, and then calculation of the level of backbone amide exchange. Tools have become available that address some of these issues, but lack sufficient integration, functionality, and accessibility required to address the needs of the H/D exchange community. To date there is no software for the analysis of H/D exchange data that comprehensively addresses these issues.
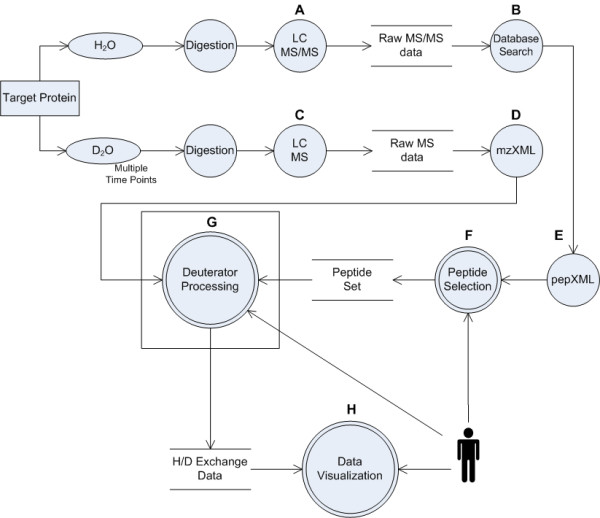
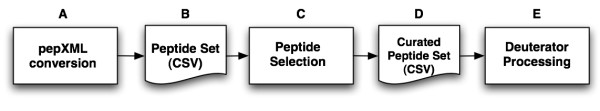
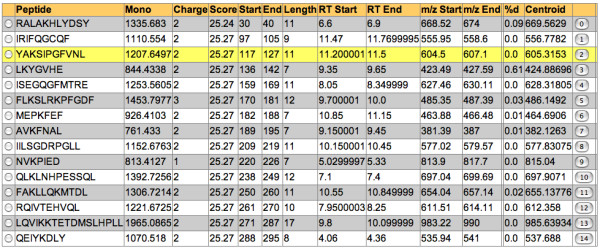
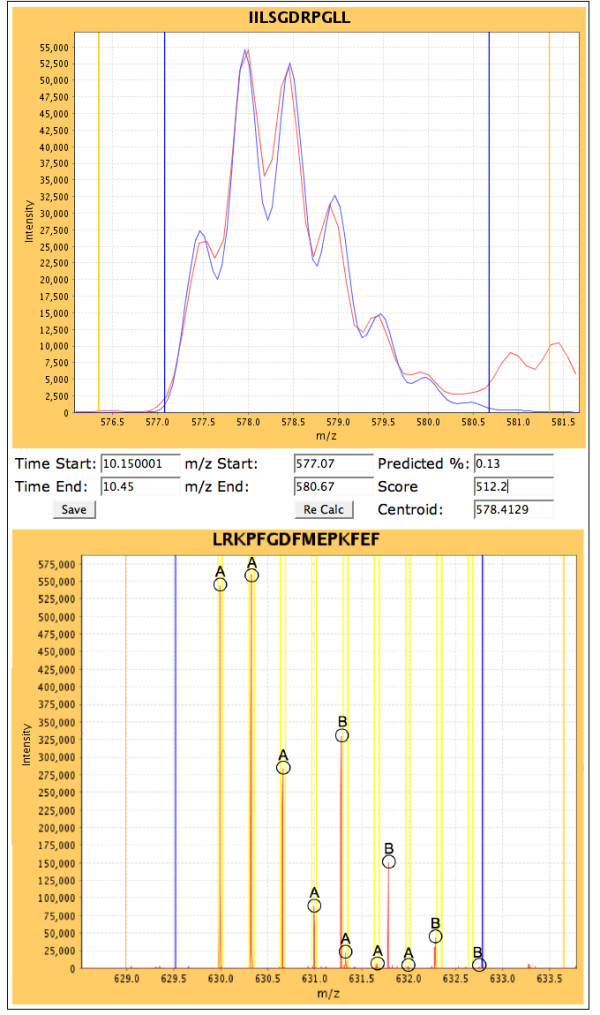
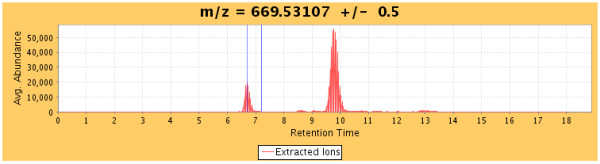
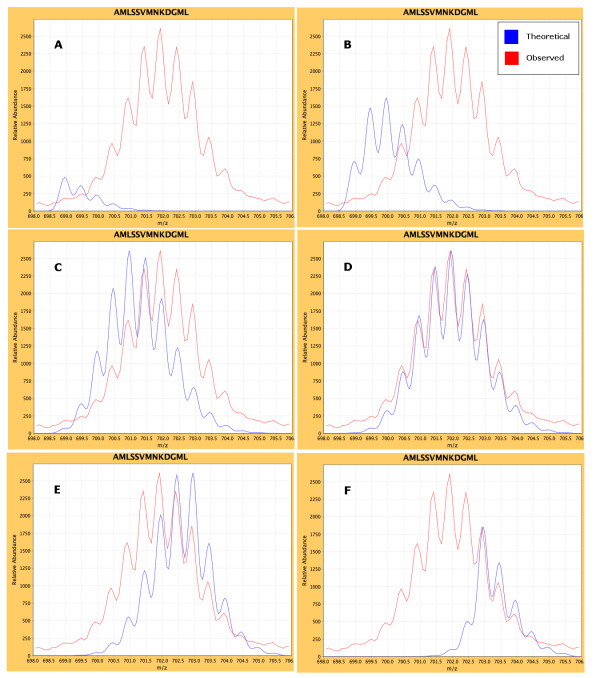
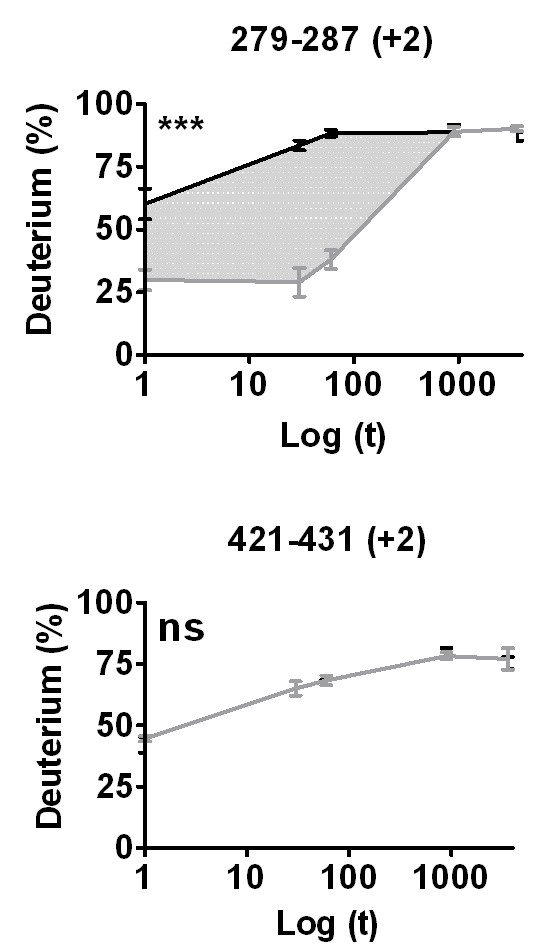
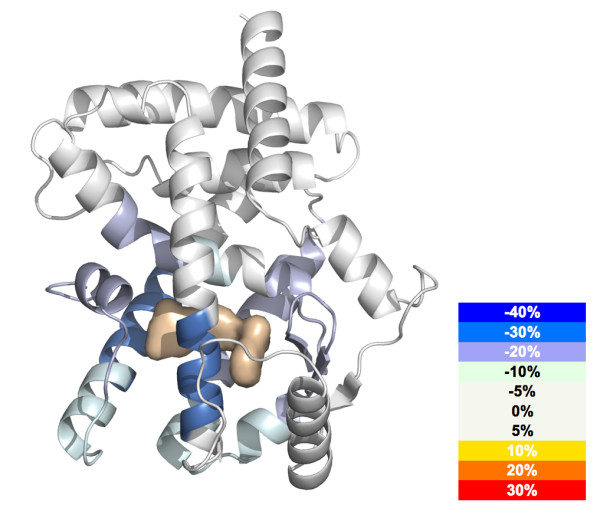
Results: We have developed an integrated software system for the automated analysis and representation of H/D exchange data that has been titled "The Deuterator". Novel approaches have been implemented that enable high throughput analysis, automated determination of deuterium incorporation, and deconvolution of overlapping peptides. This has been achieved by using methods involving iterative theoretical envelope fitting, and consideration of peak data within expected m/z ranges. Existing common file formats have been leveraged to allow compatibility with the output from the myriad of MS instrument platforms and peptide sequence database search engines.A web-based interface is used to integrate the components of The Deuterator that are able to analyze and present mass spectral data from instruments with varying resolving powers. The results, if necessary, can then be confirmed, adjusted, re-calculated and saved. Additional tools synchronize the curated calculation parameters with replicate time points, increasing throughput. Saved results can then be used to plot deuterium buildup curves and 3D structural overlays. The system has been used successfully in a production environment for over one year and is freely available as a web tool at the project home page http://deuterator.florida.scripps.edu.
Conclusion: The automated calculation and presentation of H/D exchange data in a user interface enables scientists to organize and analyze data efficiently. Integration of the different components of The Deuterator coupled with the flexibility of common data file formats allow this system to be accessible to the broadening H/D exchange community.
Figures
References
-
- Engen JR, Smith DL. Investigating protein structure and dynamics by hydrogen exchange MS. Analytical Chemistry. 2001;73:256A–265A. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
