

Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans
- PMID: 17509612
- PMCID: PMC4318564
- DOI: 10.1016/j.yjmcc.2007.04.006
Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans
Abstract
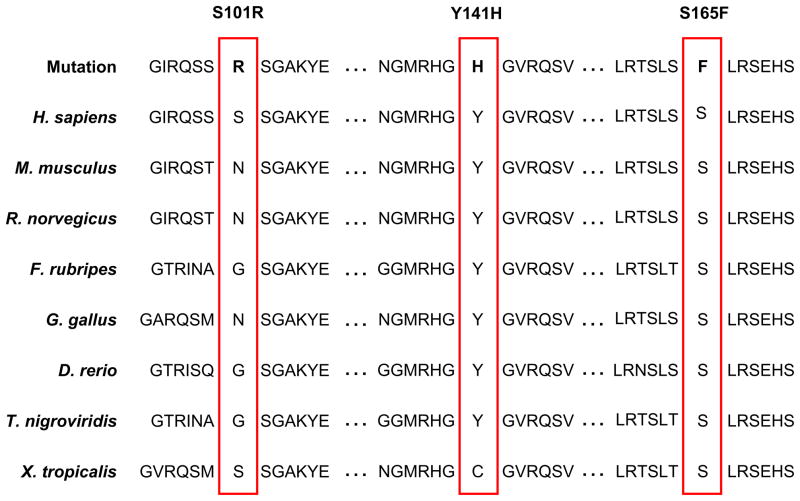
Junctophilin-2 (JPH2) is a cardiac specific member of the junctophilins, a newly characterized family of junctional membrane complex proteins important in physically approximating the plasmalemmal L-type calcium channel and the sarcoplasmic reticulum ryanodine receptor for calcium-induced calcium release. JPH2 knockout mice showed disrupted calcium transients, altered junctional membrane complex formation, cardiomyopathy, and embryonic lethality. Furthermore, JPH2 gene expression is down-regulated in murine cardiomyopathy models. To this end, we explored JPH2 as a novel candidate gene for the pathogenesis of hypertrophic cardiomyopathy (HCM) in humans. Using polymerase chain reaction, denaturing high performance liquid chromatography, and direct DNA sequencing, comprehensive open reading frame/splice site mutational analysis of JPH2 was performed on DNA obtained from 388 unrelated patients with HCM. HCM-associated JPH2 mutations were engineered and functionally characterized using immunocytochemistry, cell morphometry measurements, and live cell confocal calcium imaging. Three novel HCM-susceptibility mutations: S101R, Y141H and S165F, which localize to key functional domains, were discovered in 3/388 unrelated patients with HCM and were absent in 1000 ethnic-matched reference alleles. Functionally, each human mutation caused (i) protein reorganization of junctophilin-2, (ii) perturbations in intracellular calcium signaling, and (iii) marked cardiomyocyte hyperplasia. The molecular and functional evidence implicates defective junctophilin-2 and disrupted calcium signaling as a novel pathogenic mechanism for HCM and establishes HCM as the first human disease associated with genetic defects in JPH2. Whether susceptibility for other cardiomyopathies, such as dilated cardiomyopathy, can be conferred by mutations in JPH2 warrants investigation.
Conflict of interest statement
Figures



References
-
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–9. - PubMed
-
- Maron BJ. Hypertrophic cardiomyopathy: A systematic review. JAMA. 2002;287:1308–20. - PubMed
-
- Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, et al. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–12. - PubMed
-
- Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg H, McKenna W, Seidman CE, et al. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. - PubMed
-
- Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, et al. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:434–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA095739/CA/NCI NIH HHS/United States
- R01 HL125918/HL/NHLBI NIH HHS/United States
- R01 HL091947/HL/NHLBI NIH HHS/United States
- HD 42569/HD/NICHD NIH HHS/United States
- P01 HL094291/HL/NHLBI NIH HHS/United States
- CA 95739/CA/NCI NIH HHS/United States
- HL 69000/HL/NHLBI NIH HHS/United States
- R01 AG015556/AG/NIA NIH HHS/United States
- R01 HL069000/HL/NHLBI NIH HHS/United States
- AG 15556/AG/NIA NIH HHS/United States
- R01 HL089598/HL/NHLBI NIH HHS/United States
- R01 HD042569/HD/NICHD NIH HHS/United States
- T32 GM072474/GM/NIGMS NIH HHS/United States
- R01 HL117641/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
