

Influence of XPB helicase on recruitment and redistribution of nucleotide excision repair proteins at sites of UV-induced DNA damage
- PMID: 17509950
- PMCID: PMC3471374
- DOI: 10.1016/j.dnarep.2007.03.025
Influence of XPB helicase on recruitment and redistribution of nucleotide excision repair proteins at sites of UV-induced DNA damage
Abstract
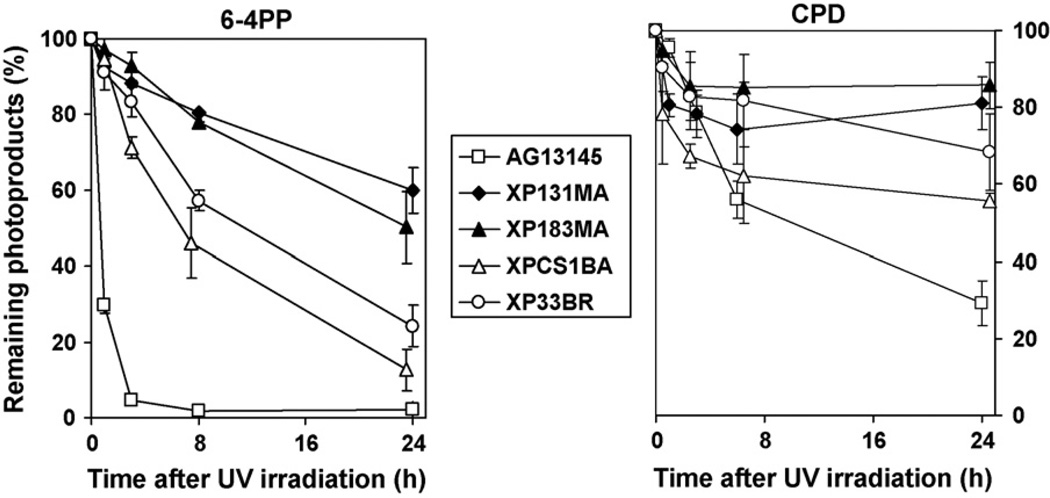
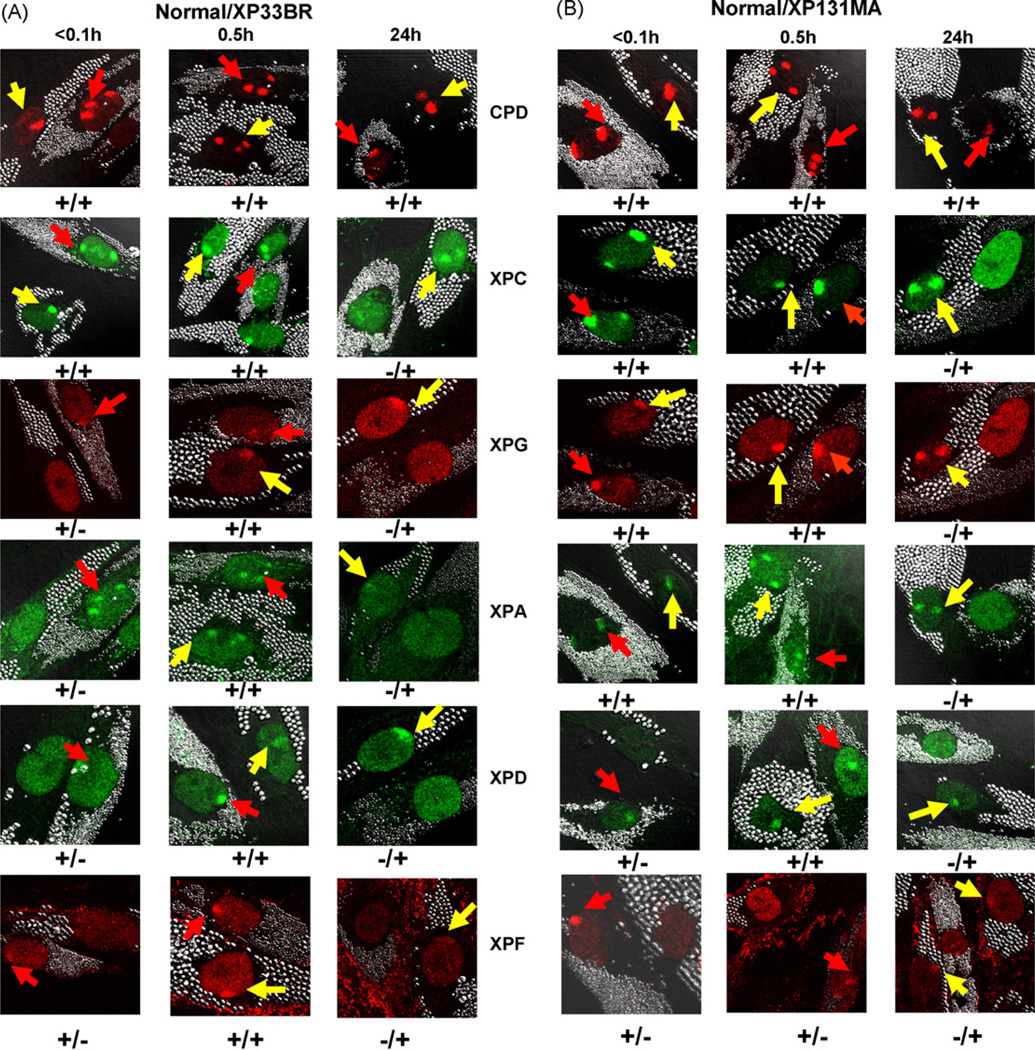
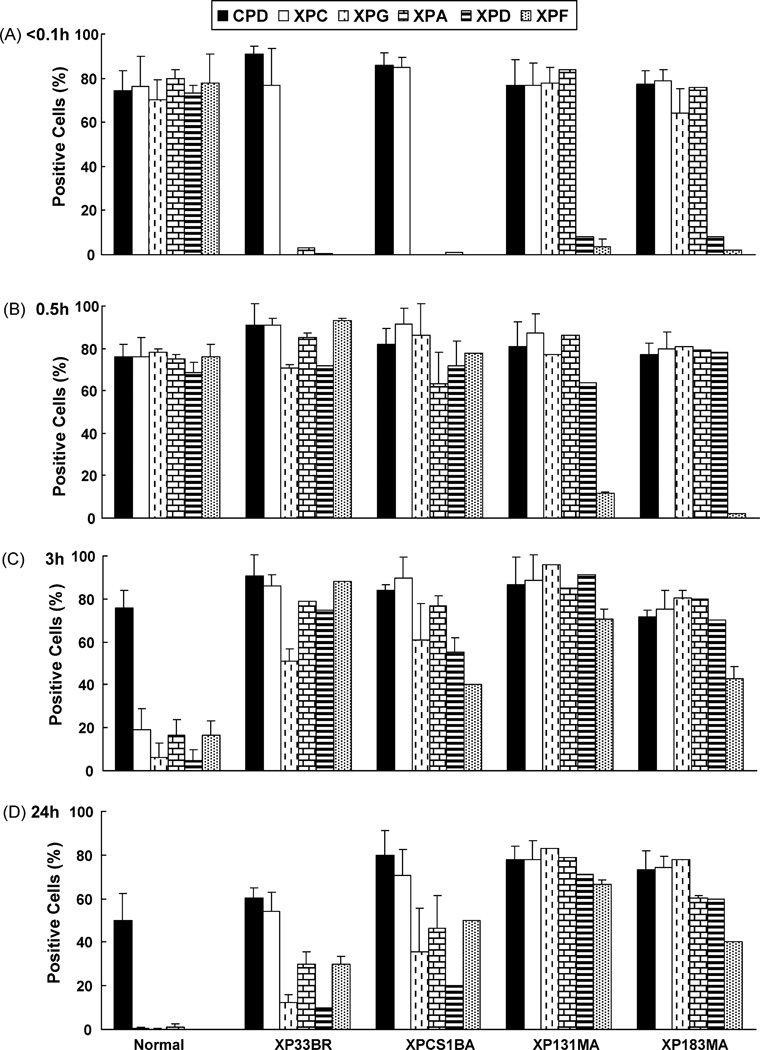
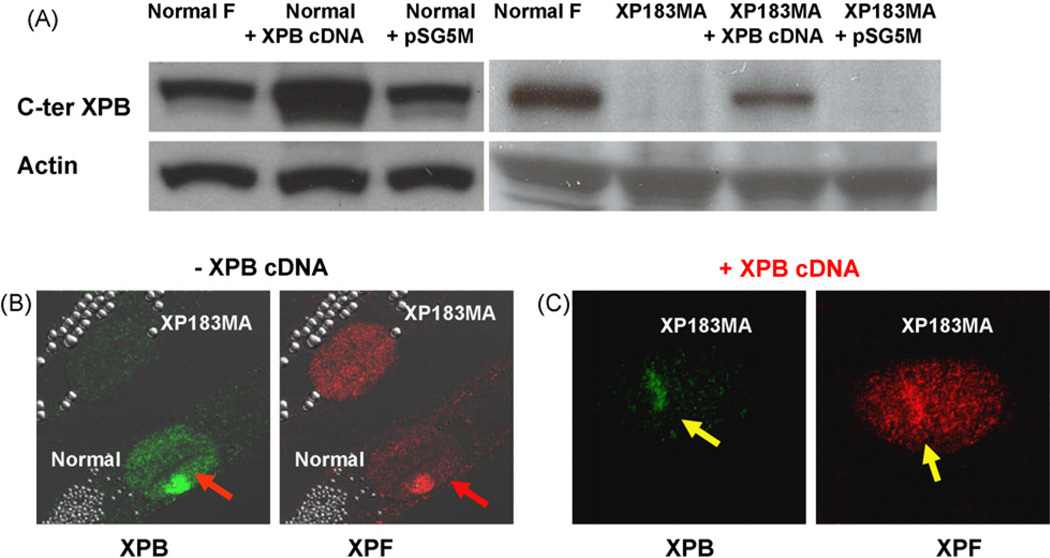
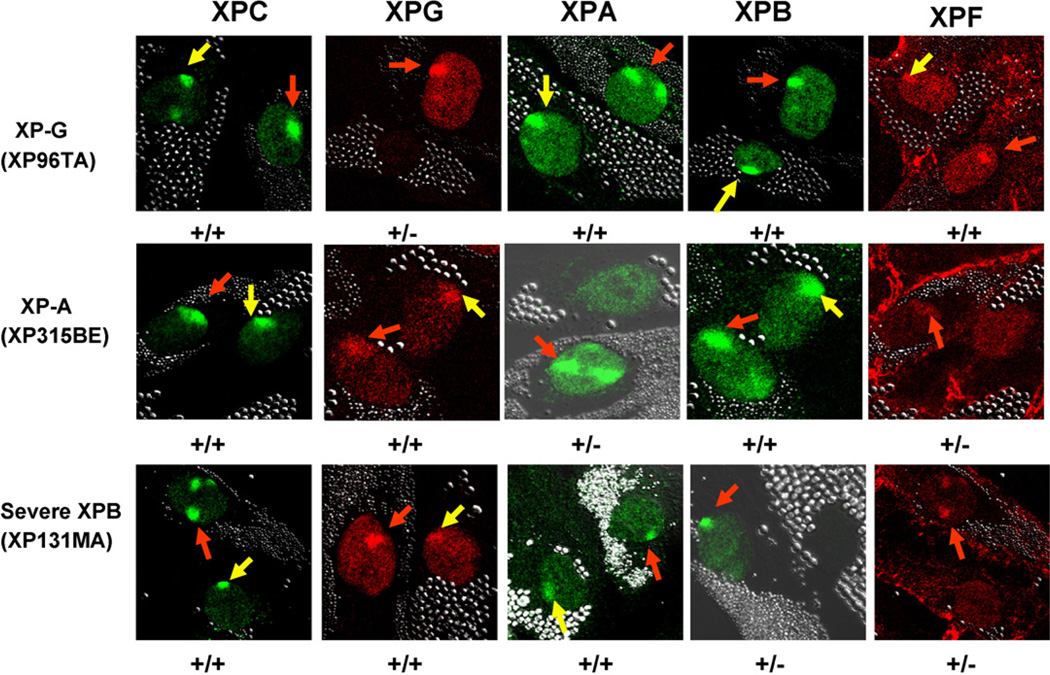
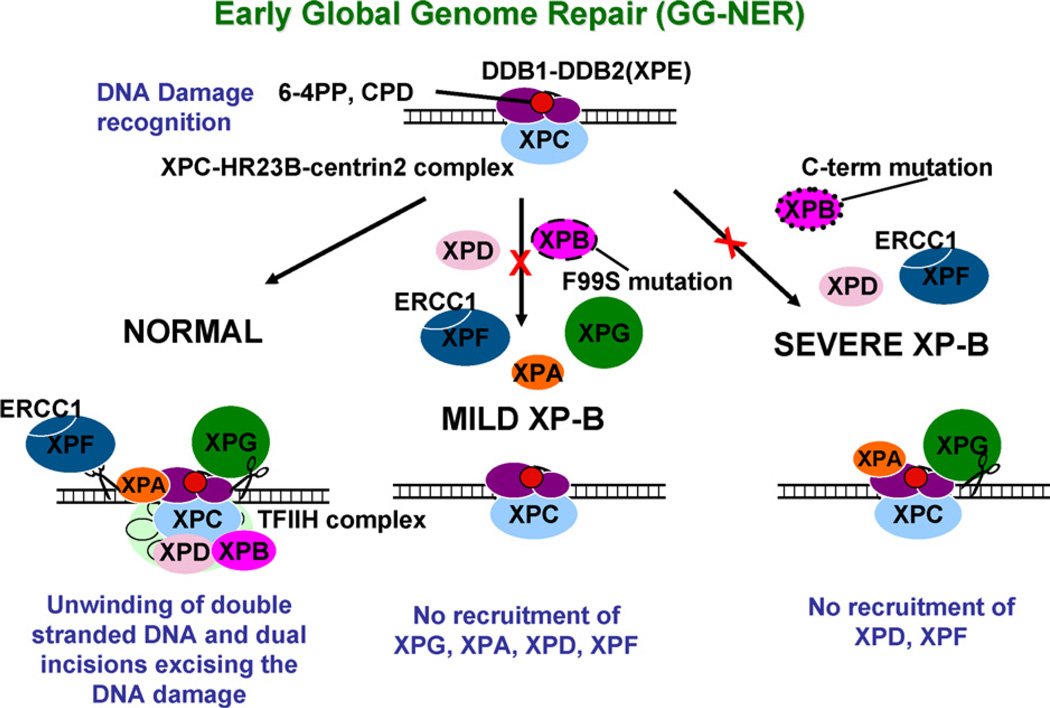
The XPB DNA helicase, a subunit of the basal transcription factor TFIIH, is also involved in nucleotide excision repair (NER). We examined recruitment of NER proteins in XP-B cells from patients with mild or severe xeroderma pigmentosum (XP) having different XPB mutations using local UV-irradiation through filters with 5 microm pores combined with fluorescent antibody labeling. XPC was rapidly recruited to UV damage sites containing DNA photoproducts (cyclobutane pyrimidine dimers, CPD) in all the XP-B and normal cells, thus reflecting its role in damage recognition prior to the function of XPB. Cells from the mild XP-B patients, with a missense mutation, showed delayed recruitment of all NER proteins except XPC to UV damage sites, demonstrating that this mutation impaired localization of these proteins. Surprisingly, in cells from severely affected patients, with a C-terminal XPB mutation, XPG and XPA proteins were normally recruited to UV damage sites demonstrating that this mutation permits recruitment of XPG and XPA. In marked contrast, in all the XP-B cells recruitment of XPF was absent immediately after UV and was delayed by 0.5 and 3 h in cells from the mild and severely affected XP patients, respectively. Redistribution of NER proteins was nearly complete in normal cells by 3 h but by 24 h redistribution was only partially present in cells from mild patients and virtually absent in cells from the severely affected patients. Ineffectual repair of UV-induced photoproducts resulting from delayed recruitment and impaired redistribution of NER proteins may contribute to the markedly increased frequency of skin cancer in XP patients.
Figures
Similar articles
-
Lack of CAK complex accumulation at DNA damage sites in XP-B and XP-B/CS fibroblasts reveals differential regulation of CAK anchoring to core TFIIH by XPB and XPD helicases during nucleotide excision repair.DNA Repair (Amst). 2012 Dec 1;11(12):942-50. doi: 10.1016/j.dnarep.2012.09.003. Epub 2012 Oct 17. DNA Repair (Amst). 2012. PMID: 23083890 Free PMC article.
-
Persistence of repair proteins at unrepaired DNA damage distinguishes diseases with ERCC2 (XPD) mutations: cancer-prone xeroderma pigmentosum vs. non-cancer-prone trichothiodystrophy.Hum Mutat. 2008 Oct;29(10):1194-208. doi: 10.1002/humu.20768. Hum Mutat. 2008. PMID: 18470933 Free PMC article.
-
Nucleotide excision repair proteins rapidly accumulate but fail to persist in human XP-E (DDB2 mutant) cells.Photochem Photobiol. 2011 May-Jun;87(3):729-33. doi: 10.1111/j.1751-1097.2011.00909.x. Epub 2011 Mar 9. Photochem Photobiol. 2011. PMID: 21388382 Free PMC article.
-
Xeroderma pigmentosum and molecular cloning of DNA repair genes.Anticancer Res. 1996 Mar-Apr;16(2):693-708. Anticancer Res. 1996. PMID: 8687116 Review.
-
XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase.DNA Repair (Amst). 2011 Jul 15;10(7):697-713. doi: 10.1016/j.dnarep.2011.04.028. Epub 2011 May 14. DNA Repair (Amst). 2011. PMID: 21571596 Free PMC article. Review.
Cited by
-
The NR4A2 nuclear receptor is recruited to novel nuclear foci in response to UV irradiation and participates in nucleotide excision repair.PLoS One. 2013 Nov 6;8(11):e78075. doi: 10.1371/journal.pone.0078075. eCollection 2013. PLoS One. 2013. PMID: 24223135 Free PMC article.
-
UV-induced histone H2AX phosphorylation and DNA damage related proteins accumulate and persist in nucleotide excision repair-deficient XP-B cells.DNA Repair (Amst). 2011 Jan 2;10(1):5-15. doi: 10.1016/j.dnarep.2010.09.004. Epub 2010 Oct 13. DNA Repair (Amst). 2011. PMID: 20947453 Free PMC article.
-
Lack of CAK complex accumulation at DNA damage sites in XP-B and XP-B/CS fibroblasts reveals differential regulation of CAK anchoring to core TFIIH by XPB and XPD helicases during nucleotide excision repair.DNA Repair (Amst). 2012 Dec 1;11(12):942-50. doi: 10.1016/j.dnarep.2012.09.003. Epub 2012 Oct 17. DNA Repair (Amst). 2012. PMID: 23083890 Free PMC article.
-
Telomere attrition and genomic instability in xeroderma pigmentosum type-b deficient fibroblasts under oxidative stress.J Cell Mol Med. 2010 Jan;14(1-2):403-16. doi: 10.1111/j.1582-4934.2009.00945.x. Epub 2009 Oct 16. J Cell Mol Med. 2010. PMID: 19840190 Free PMC article.
-
Dissection of the molecular defects caused by pathogenic mutations in the DNA repair factor XPC.Mol Cell Biol. 2008 Dec;28(23):7225-35. doi: 10.1128/MCB.00781-08. Epub 2008 Sep 22. Mol Cell Biol. 2008. PMID: 18809580 Free PMC article.
References
-
- Bootsma D, Kraemer KH, Cleaver JE, Hoeijmakers JHJ. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler KW, editors. The Genetic Basis of Human Cancer. 2nd ed. New York: McGraw-Hill; 2002. pp. 211–237.
-
- Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. - PubMed
-
- Moriwaki S, Kraemer KH. Xeroderma pigmentosum—bridging a gap between clinic and laboratory. Photodermatol. Photoimmunol. Photomed. 2001;17:47–54. - PubMed
-
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. Washington, DC: ASM Press; 2006.
-
- Petit C, Sancar A. Nucleotide excision repair: from E. coli to man. Biochimie. 1999;81:15–25. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
