

The origin of modern metabolic networks inferred from phylogenomic analysis of protein architecture
- PMID: 17517598
- PMCID: PMC1890499
- DOI: 10.1073/pnas.0701214104
The origin of modern metabolic networks inferred from phylogenomic analysis of protein architecture
Abstract
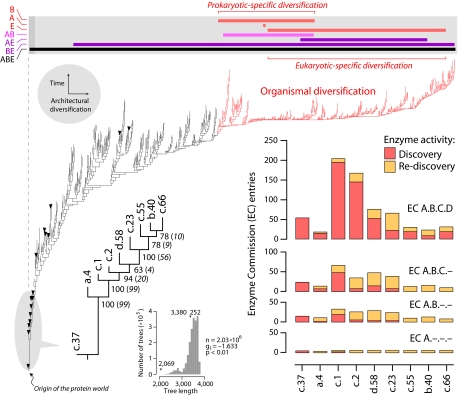
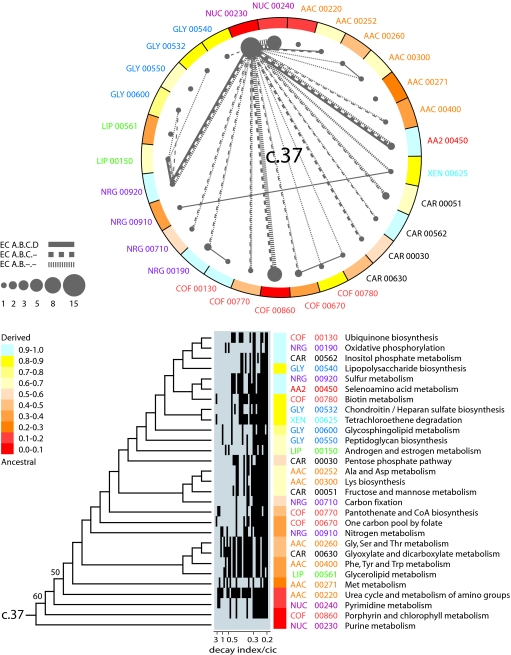
Metabolism represents a complex collection of enzymatic reactions and transport processes that convert metabolites into molecules capable of supporting cellular life. Here we explore the origins and evolution of modern metabolism. Using phylogenomic information linked to the structure of metabolic enzymes, we sort out recruitment processes and discover that most enzymatic activities were associated with the nine most ancient and widely distributed protein fold architectures. An analysis of newly discovered functions showed enzymatic diversification occurred early, during the onset of the modern protein world. Most importantly, phylogenetic reconstruction exercises and other evidence suggest strongly that metabolism originated in enzymes with the P-loop hydrolase fold in nucleotide metabolism, probably in pathways linked to the purine metabolic subnetwork. Consequently, the first enzymatic takeover of an ancient biochemistry or prebiotic chemistry was related to the synthesis of nucleotides for the RNA world.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Barabási AL, Oltvai ZN. Nat Rev Genet. 2004;5:101–113. - PubMed
-
- Schmidt S, Sunyaev S, Bork P, Dandekar T. Trends Biochem Sci. 2003;28:336–341. - PubMed
-
- Ycas M. J Theor Biol. 1974;44:145–160. - PubMed
-
- Jensen RA. Annu Rev Microbiol. 1976;30:409–425. - PubMed
-
- Copley RR, Bork P. J Mol Biol. 2000;303:627–640. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
