

Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains
- PMID: 17517971
- PMCID: PMC2118613
- DOI: 10.1084/jem.20070301
Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains
Abstract
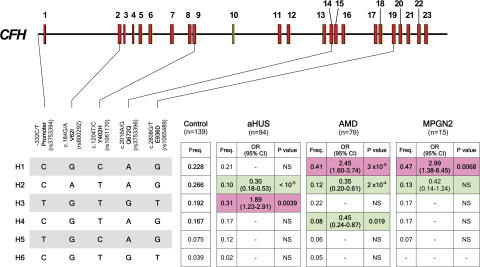
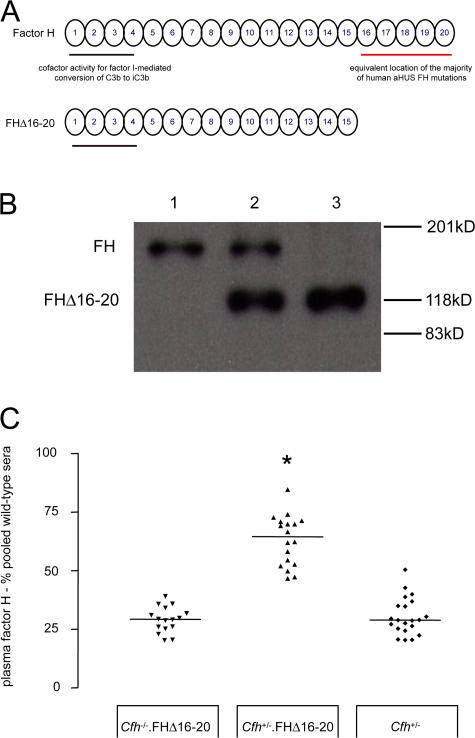
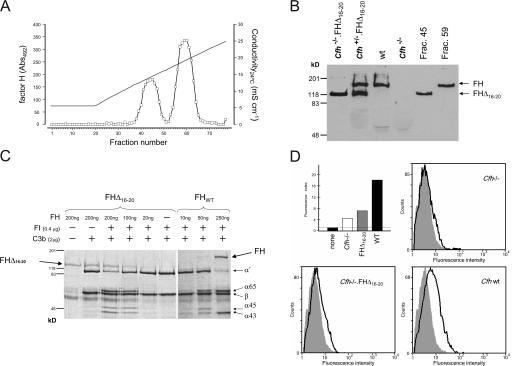
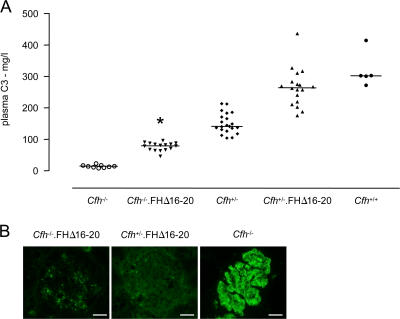
Factor H (FH) is an abundant serum glycoprotein that regulates the alternative pathway of complement-preventing uncontrolled plasma C3 activation and nonspecific damage to host tissues. Age-related macular degeneration (AMD), atypical hemolytic uremic syndrome (aHUS), and membranoproliferative glomerulonephritis type II (MPGN2) are associated with polymorphisms or mutations in the FH gene (Cfh), suggesting the existence of a genotype-phenotype relationship. Although AMD and MPGN2 share pathological similarities with the accumulation of complement-containing debris within the eye and kidney, respectively, aHUS is characterized by renal endothelial injury. This pathological distinction was reflected in our Cfh association analysis, which demonstrated that although AMD and MPGN2 share a Cfh at-risk haplotype, the haplotype for aHUS was unique. FH-deficient mice have uncontrolled plasma C3 activation and spontaneously develop MPGN2 but not aHUS. We show that these mice, transgenically expressing a mouse FH protein functionally equivalent to aHUS-associated human FH mutants, regulate C3 activation in plasma and spontaneously develop aHUS but not MPGN2. These animals represent the first model of aHUS and provide in vivo evidence that effective plasma C3 regulation and the defective control of complement activation on renal endothelium are the critical events in the molecular pathogenesis of FH-associated aHUS.
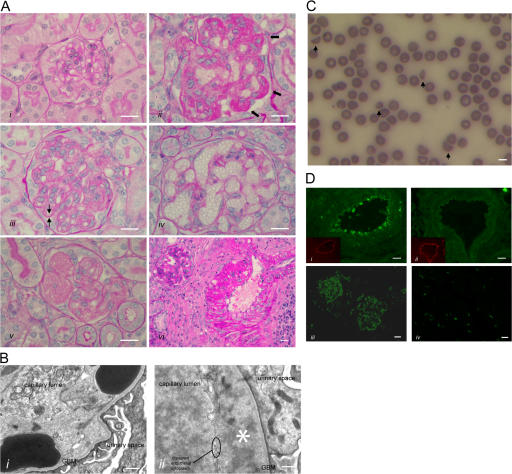
Figures
References
-
- Caprioli, J., P. Bettinaglio, P.F. Zipfel, B. Amadei, E. Daina, S. Gamba, C. Skerka, N. Marziliano, G. Remuzzi, and M. Noris. 2001. The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J. Am. Soc. Nephrol. 12:297–307. - PubMed
-
- Warwicker, P., T.H. Goodship, R.L. Donne, Y. Pirson, A. Nicholls, R.M. Ward, P. Turnpenny, and J.A. Goodship. 1998. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 53:836–844. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
