

The neurobiologist's guide to structural biology: a primer on why macromolecular structure matters and how to evaluate structural data
- PMID: 17521566
- PMCID: PMC3011226
- DOI: 10.1016/j.neuron.2007.04.026
The neurobiologist's guide to structural biology: a primer on why macromolecular structure matters and how to evaluate structural data
Abstract
Structural biology now plays a prominent role in addressing questions central to understanding how excitable cells function. Although interest in the insights gained from the definition and dissection of macromolecular anatomy is high, many neurobiologists remain unfamiliar with the methods employed. This primer aims to help neurobiologists understand approaches for probing macromolecular structure and where the limits and challenges remain. Using examples of macromolecules with neurobiological importance, the review covers X-ray crystallography, electron microscopy (EM), small-angle X-ray scattering (SAXS), and nuclear magnetic resonance (NMR) and biophysical methods with which these approaches are often paired: isothermal titration calorimetry (ITC), equilibrium analytical ultracentifugation, and molecular dynamics (MD).
Figures
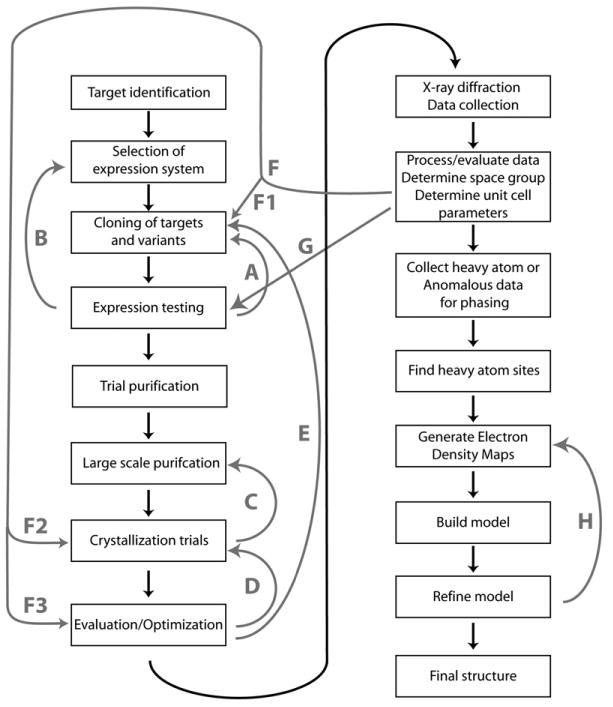
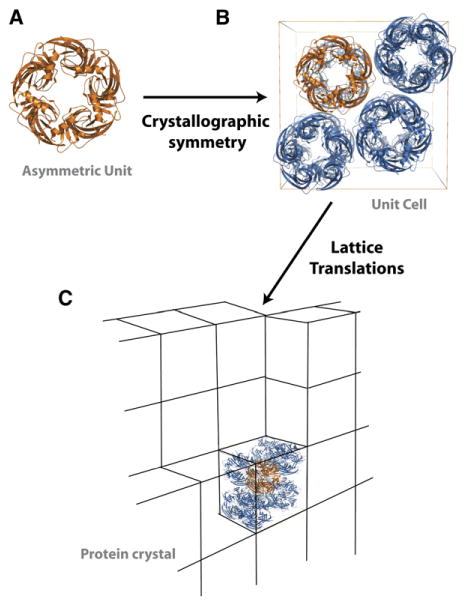
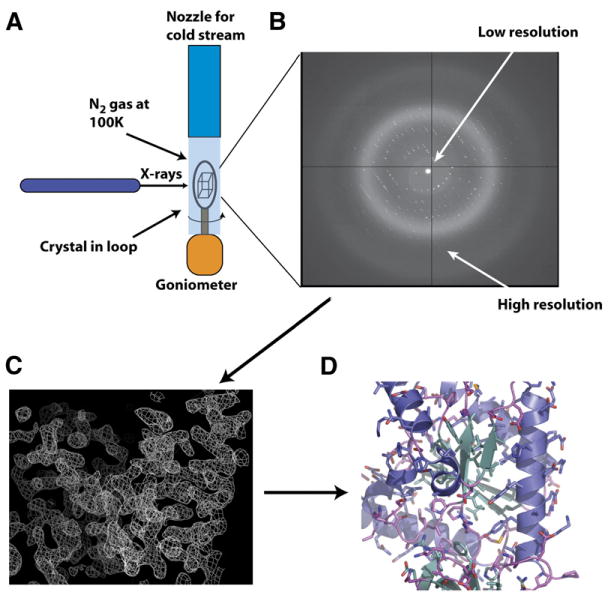
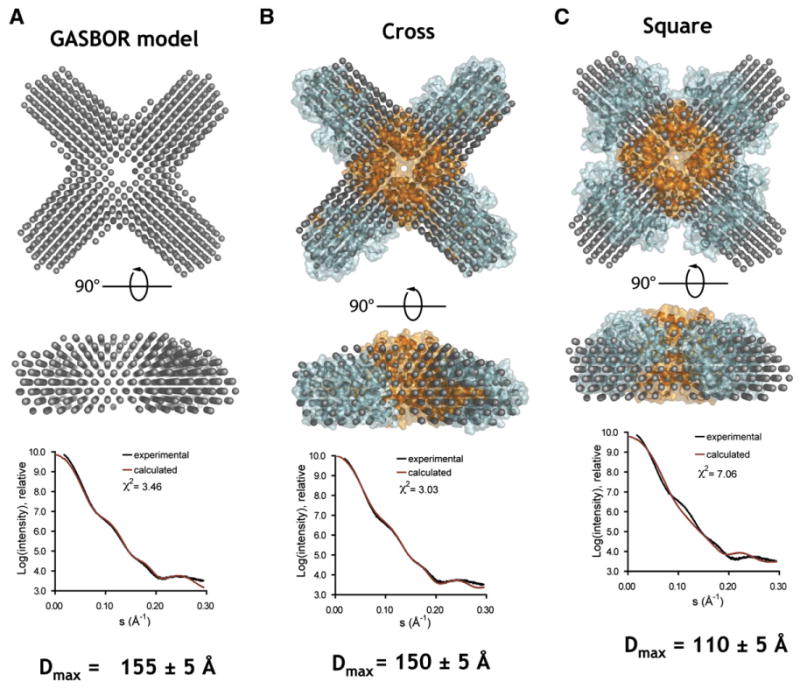
Similar articles
-
Integrative Approaches in Structural Biology: A More Complete Picture from the Combination of Individual Techniques.Biomolecules. 2019 Aug 14;9(8):370. doi: 10.3390/biom9080370. Biomolecules. 2019. PMID: 31416261 Free PMC article. Review.
-
Whaddaya Know: A Guide to Uncertainty and Subjectivity in Structural Biology.Trends Biochem Sci. 2017 Feb;42(2):155-167. doi: 10.1016/j.tibs.2016.11.002. Epub 2017 Jan 12. Trends Biochem Sci. 2017. PMID: 28089412 Review.
-
Biophysical applications in structural and molecular biology.Biol Chem. 2021 Jul 7;402(10):1155-1177. doi: 10.1515/hsz-2021-0232. Print 2021 Sep 27. Biol Chem. 2021. PMID: 34218543 Review.
-
Direct imaging electron microscopy (EM) methods in modern structural biology: overview and comparison with X-ray crystallography and single-particle cryo-EM reconstruction in the studies of large macromolecules.Biol Cell. 2014 Oct;106(10):323-45. doi: 10.1111/boc.201300081. Epub 2014 Aug 27. Biol Cell. 2014. PMID: 25040059 Review.
-
Methods for Crystallization and Structural Determination of M-T7 Protein from Myxoma Virus.Methods Mol Biol. 2021;2225:125-162. doi: 10.1007/978-1-0716-1012-1_8. Methods Mol Biol. 2021. PMID: 33108661
Cited by
-
Structure of a Ca(2+)/CaM:Kv7.4 (KCNQ4) B-helix complex provides insight into M current modulation.J Mol Biol. 2013 Jan 23;425(2):378-94. doi: 10.1016/j.jmb.2012.11.023. Epub 2012 Nov 23. J Mol Biol. 2013. PMID: 23178170 Free PMC article.
-
Conduits of life's spark: a perspective on ion channel research since the birth of neuron.Neuron. 2013 Oct 30;80(3):658-74. doi: 10.1016/j.neuron.2013.10.040. Neuron. 2013. PMID: 24183018 Free PMC article. Review.
-
Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation.Channels (Austin). 2010 Nov-Dec;4(6):459-74. doi: 10.4161/chan.4.6.12867. Channels (Austin). 2010. PMID: 21139419 Free PMC article. Review.
-
Molecular modeling of the full-length human TRPV1 channel in closed and desensitized states.J Membr Biol. 2008 Jun;223(3):161-72. doi: 10.1007/s00232-008-9123-7. Epub 2008 Sep 14. J Membr Biol. 2008. PMID: 18791833
-
Different ligands of the TRPV3 cation channel cause distinct conformational changes as revealed by intrinsic tryptophan fluorescence quenching.J Biol Chem. 2015 May 15;290(20):12964-74. doi: 10.1074/jbc.M114.628925. Epub 2015 Mar 31. J Biol Chem. 2015. PMID: 25829496 Free PMC article.
References
-
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301:610–615. - PubMed
-
- Aricescu AR, Lu W, Jones EY. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr D Biol Crystallogr. 2006b;62:1243–1250. - PubMed
-
- Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. - PubMed
-
- Armstrong N, Sun Y, Chen GQ, Gouaux E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 1998;395:913–917. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
