

Tuning immune tolerance with vasoactive intestinal peptide: a new therapeutic approach for immune disorders
- PMID: 17521775
- PMCID: PMC2071927
- DOI: 10.1016/j.peptides.2007.04.008
Tuning immune tolerance with vasoactive intestinal peptide: a new therapeutic approach for immune disorders
Abstract
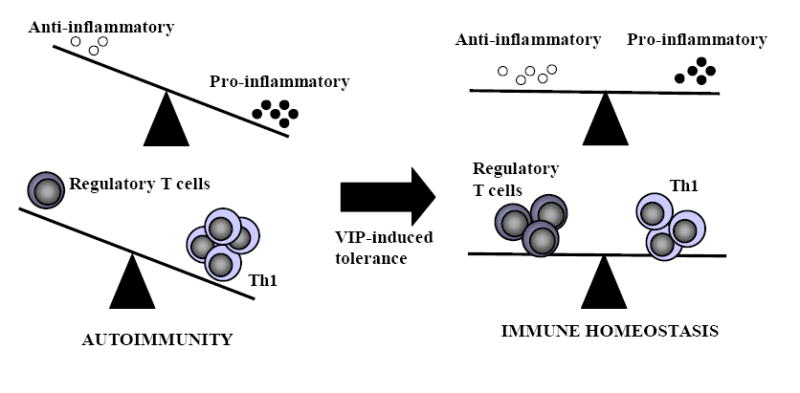
The induction of immune tolerance is essential for the maintenance of immune homeostasis and to limit the occurrence of exacerbated inflammatory and autoimmune conditions. Multiple mechanisms act together to ensure self-tolerance, including central clonal deletion, cytokine deviation and induction of regulatory T cells. Identifying the factors that regulate these processes is crucial for the development of new therapies of autoimmune diseases and transplantation. The vasoactive intestinal peptide (VIP) is a well-characterized endogenous anti-inflammatory neuropeptide with therapeutic potential for a variety of immune disorders. Here, we examine the latest research findings, which indicate that VIP participates in maintaining immune tolerance in two distinct ways: by regulating the balance between pro-inflammatory and anti-inflammatory factors, and by inducing the emergence of regulatory T cells with suppressive activity against autoreactive T-cell effectors.
Figures



Similar articles
-
Emerging roles of vasoactive intestinal peptide: a new approach for autoimmune therapy.Ann Rheum Dis. 2007 Nov;66 Suppl 3(Suppl 3):iii70-6. doi: 10.1136/ard.2007.078519. Ann Rheum Dis. 2007. PMID: 17934101 Free PMC article. Review.
-
Neuropeptides as therapeutic approach to autoimmune diseases.Curr Pharm Des. 2010;16(28):3158-72. doi: 10.2174/138161210793292465. Curr Pharm Des. 2010. PMID: 20687881 Review.
-
Potential applications of vasoactive intestinal Peptide-based therapies on transplantation.Endocr Metab Immune Disord Drug Targets. 2012 Dec;12(4):333-43. doi: 10.2174/187153012803832567. Endocr Metab Immune Disord Drug Targets. 2012. PMID: 23094830 Review.
-
Endogenous anti-inflammatory neuropeptides and pro-resolving lipid mediators: a new therapeutic approach for immune disorders.J Cell Mol Med. 2008 Oct;12(5B):1830-47. doi: 10.1111/j.1582-4934.2008.00387.x. Epub 2008 Jun 28. J Cell Mol Med. 2008. PMID: 18554314 Free PMC article. Review.
-
Vasoactive intestinal peptide and regulatory T-cell induction: a new mechanism and therapeutic potential for immune homeostasis.Trends Mol Med. 2007 Jun;13(6):241-51. doi: 10.1016/j.molmed.2007.04.003. Epub 2007 Apr 27. Trends Mol Med. 2007. PMID: 17467339
Cited by
-
Negative regulators that mediate ocular immune privilege.J Leukoc Biol. 2018 Feb 12:10.1002/JLB.3MIR0817-337R. doi: 10.1002/JLB.3MIR0817-337R. Online ahead of print. J Leukoc Biol. 2018. PMID: 29431864 Free PMC article. Review.
-
Tear levels of neuropeptides increase after specific allergen challenge in allergic conjunctivitis.Mol Vis. 2011 Jan 7;17:47-52. Mol Vis. 2011. PMID: 21245958 Free PMC article.
-
The effects of vasoactive intestinal peptide in the rat model of experimental autoimmune neuritis and the implications for treatment of acute inflammatory demyelinating polyradiculoneuropathy or Guillain-Barré syndrome.Drug Des Devel Ther. 2018 Nov 6;12:3817-3824. doi: 10.2147/DDDT.S175331. eCollection 2018. Drug Des Devel Ther. 2018. PMID: 30464413 Free PMC article.
-
VIP plasma levels associate with survival in severe COVID-19 patients, correlating with protective effects in SARS-CoV-2-infected cells.J Leukoc Biol. 2022 May;111(5):1107-1121. doi: 10.1002/JLB.5COVA1121-626R. Epub 2022 Mar 24. J Leukoc Biol. 2022. PMID: 35322471 Free PMC article.
-
Regulatory dendritic cells for immunotherapy in immunologic diseases.Front Immunol. 2014 Jan 31;5:7. doi: 10.3389/fimmu.2014.00007. eCollection 2014. Front Immunol. 2014. PMID: 24550907 Free PMC article. Review.
References
-
- Abad C, Martinez C, Juarranz MG, Arranz A, Leceta J, Delgado M, et al. Therapeutic effects of vasoactive intestinal peptide in the trinitrobenzene sulfonic acid mice model of Crohn’s disease. Gastroenterology. 2003;124:961–71. - PubMed
-
- Bangale Y, Karle S, Planque S, Zhou YX, Taguchi H, Nishiyama Y, et al. VIPase autoantibodies in Fas-defective mice and patients with autoimmune disease. FASEB J. 2003;17:628–35. - PubMed
-
- Banner KH, Trevethick MA. PDE4 inhibition: a novel approach for the treatment of inflammatory bowel disease. Trends Pharmacol Sci. 2004;25:430–6. - PubMed
-
- Blalock JE. The immune system as the sixth sense. J Intern Med. 2005;257:126–38. - PubMed
-
- Bluestone JA. Regulatory T-cell therapy: is it ready for the clinic? Nat Rev Immunol. 2005;5:343–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
