

Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model
- PMID: 17522299
- PMCID: PMC6672753
- DOI: 10.1523/JNEUROSCI.1139-07.2007
Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model
Abstract
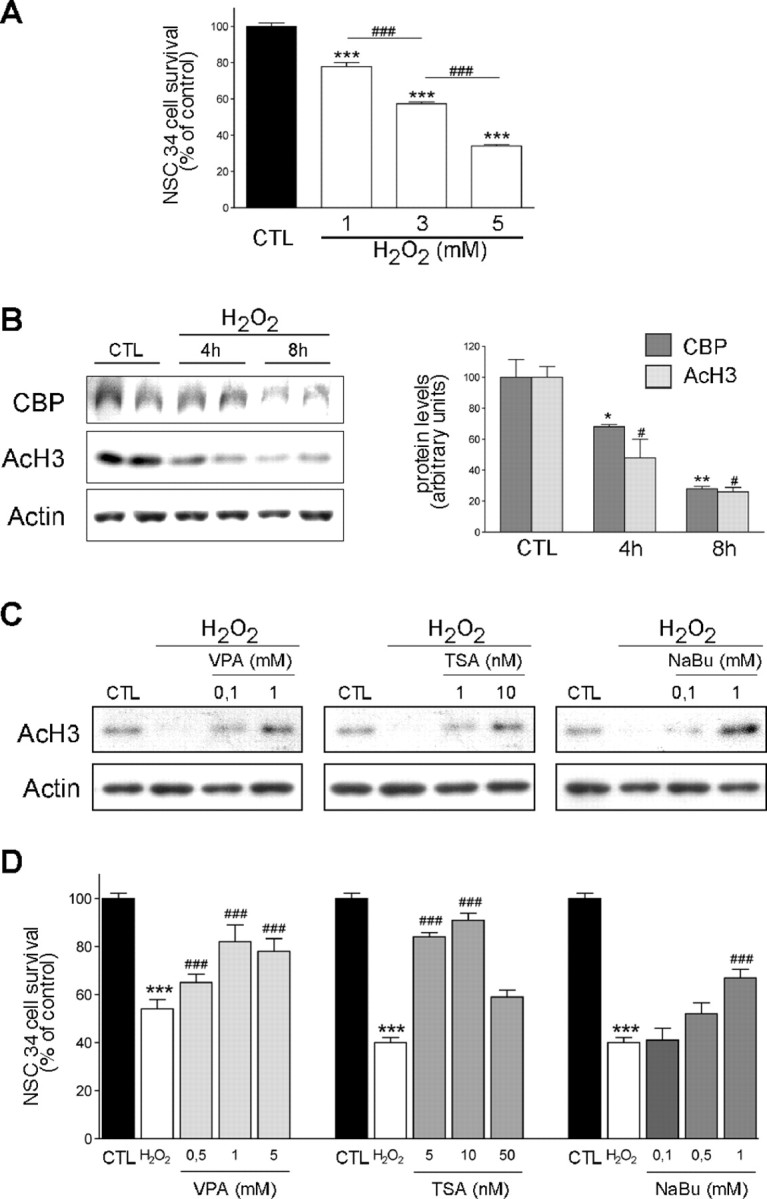
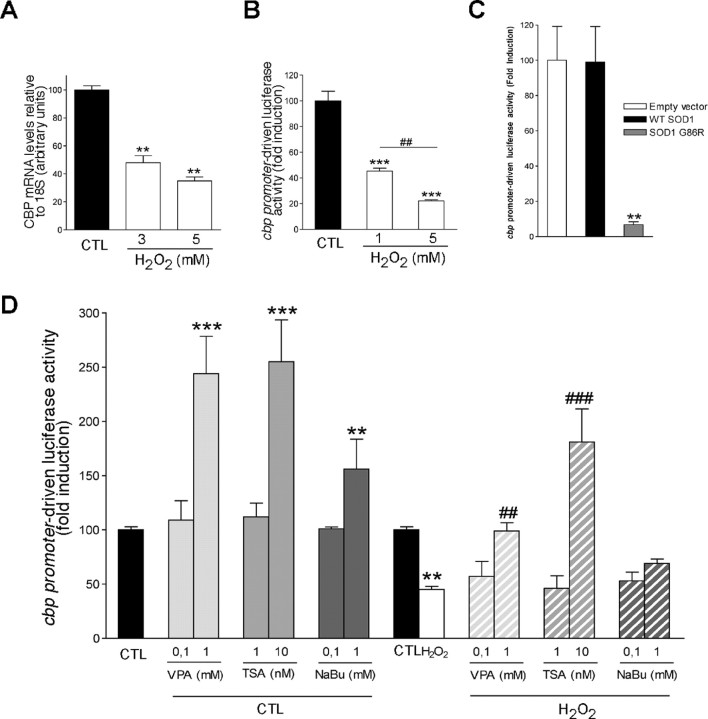
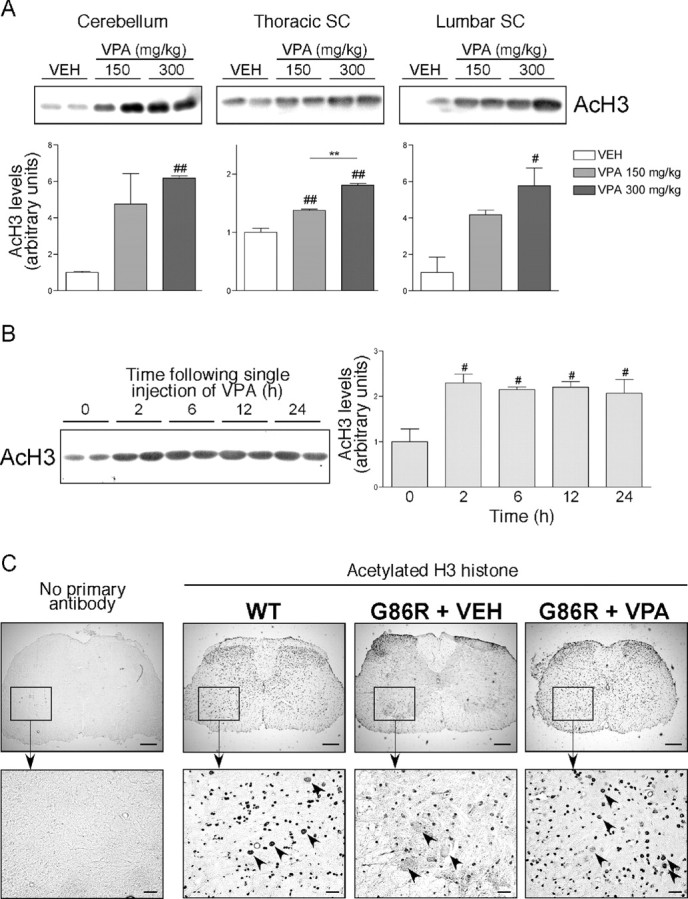
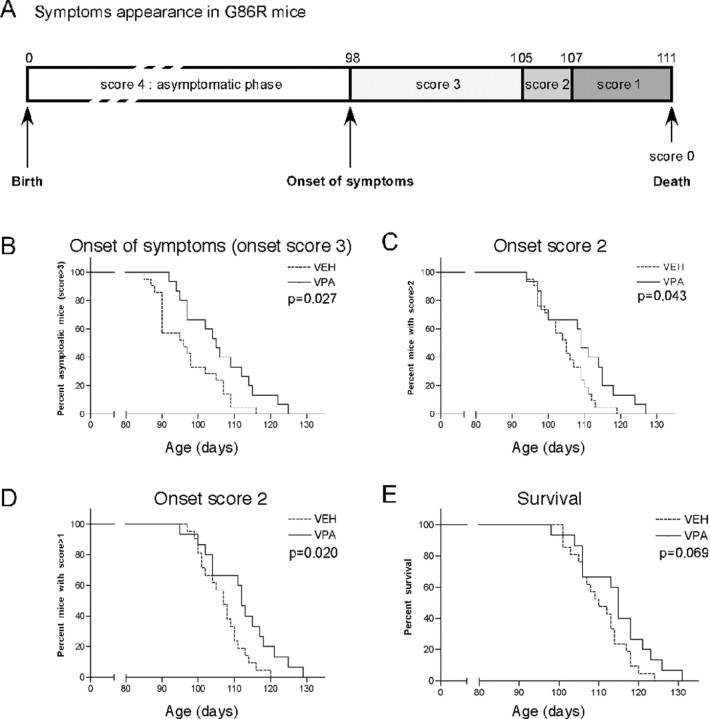
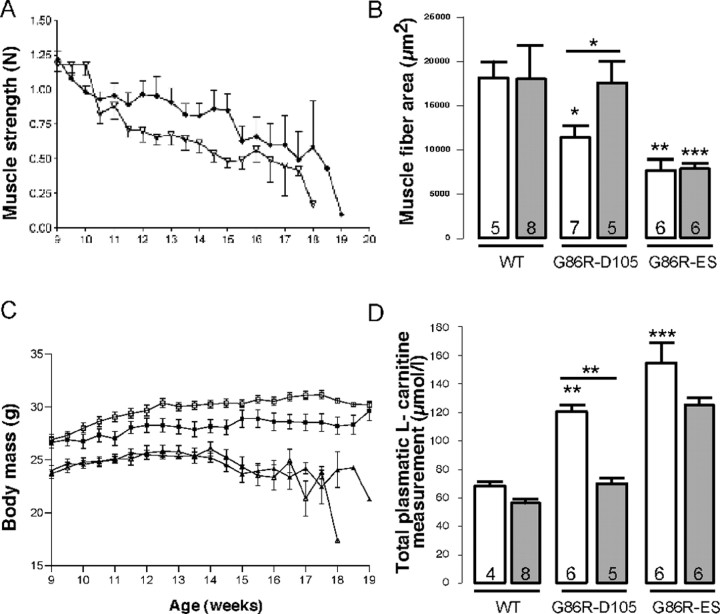
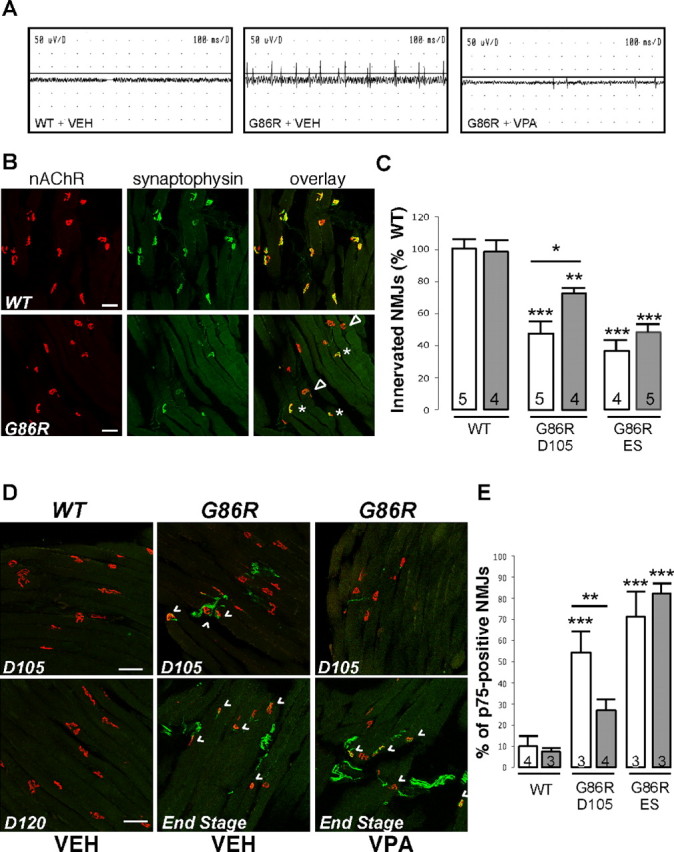
Amyotrophic lateral sclerosis (ALS) is characterized by motoneuron (MN) degeneration, generalized weakness, and muscle atrophy. The premature death of MNs is thought to be a determinant in the onset of this disease. In a transgenic mouse model of ALS expressing the G86R mutant superoxide dismutase 1 (mSOD1), we demonstrated previously that CREB (cAMP response element-binding protein)-binding protein (CBP) and histone acetylation levels were specifically decreased in nuclei of degenerating MNs. We show here that oxidative stress and mSOD1 overexpression can both impinge on CBP levels by transcriptional repression, in an MN-derived cell line. Histone deacetylase inhibitor (HDACi) treatment was able to reset proper acetylation levels and displayed an efficient neuroprotective capacity against oxidative stress in vitro. Interestingly, HDACi also upregulated CBP transcriptional expression in MNs. Moreover, when injected to G86R mice in vivo, the HDACi sodium valproate (VPA) maintained normal acetylation levels in the spinal cord, efficiently restored CBP levels in MNs, and significantly prevented MN death in these animals. However, despite neuroprotection, mean survival of treated animals was not significantly improved (<5%), and they died presenting the classical ALS symptoms. VPA was not able to prevent disruption of neuromuscular junctions, although it slightly delayed the onset of motor decline and retarded muscular atrophy to some extent. Together, these data show that neuroprotection can improve disease onset, but clearly provide evidence that one can uncouple MN survival from whole-animal survival and point to the neuromuscular junction perturbation as a primary event of ALS onset.
Figures

References
-
- Andersen PM, Sims KB, Xin WW, Kiely R, O'Neill G, Ravits J, Pioro E, Harati Y, Brower RD, Levine JS, Heinicke HU, Seltzer W, Boss M, Brown RH., Jr Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:62–73. - PubMed
-
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. - PubMed
-
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–591. - PubMed
-
- Bjornskov EK, Norris FH, Jr, Mower-Kuby J. Quantitative axon terminal and end-plate morphology in amyotrophic lateral sclerosis. Arch Neurol. 1984;41:527–530. - PubMed
-
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous