

Estrogen signaling via a linear pathway involving insulin-like growth factor I receptor, matrix metalloproteinases, and epidermal growth factor receptor to activate mitogen-activated protein kinase in MCF-7 breast cancer cells
- PMID: 17525128
- PMCID: PMC2727866
- DOI: 10.1210/en.2007-0240
Estrogen signaling via a linear pathway involving insulin-like growth factor I receptor, matrix metalloproteinases, and epidermal growth factor receptor to activate mitogen-activated protein kinase in MCF-7 breast cancer cells
Abstract
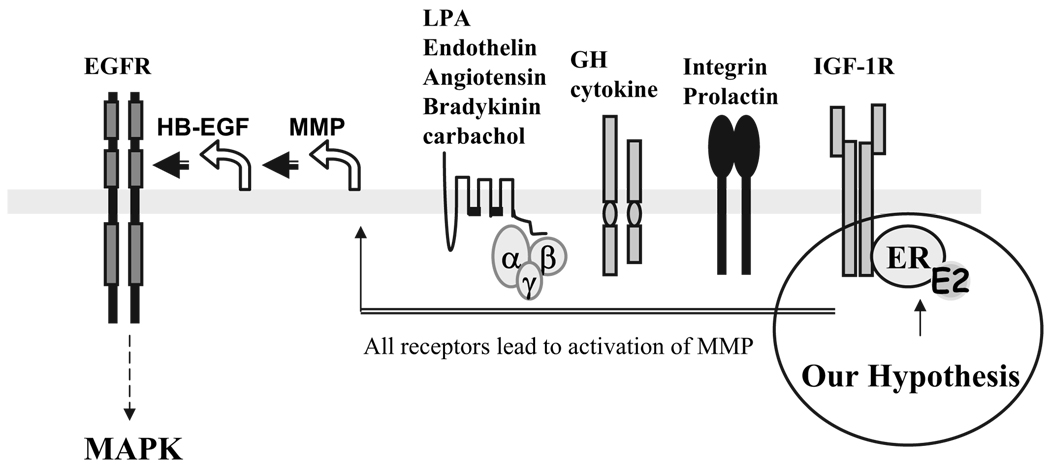
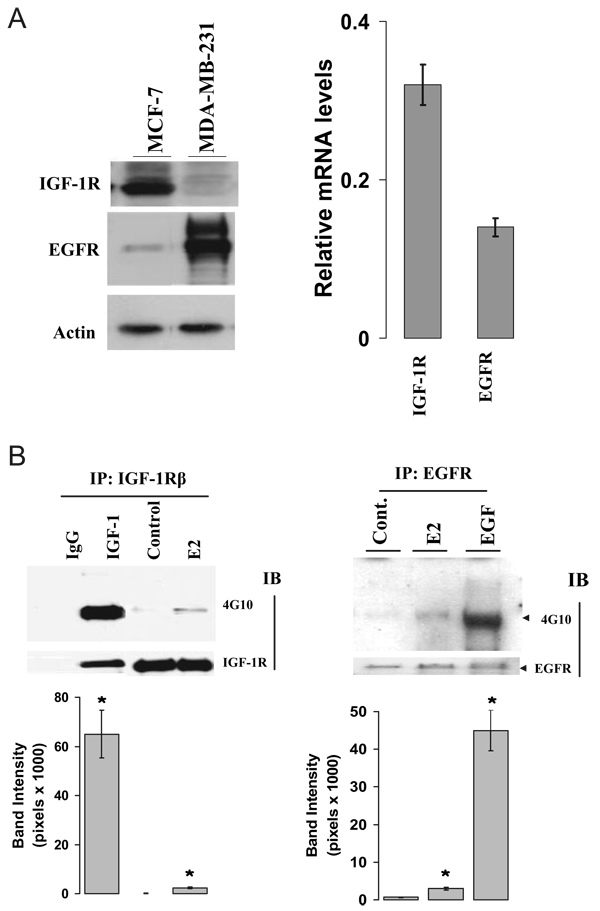
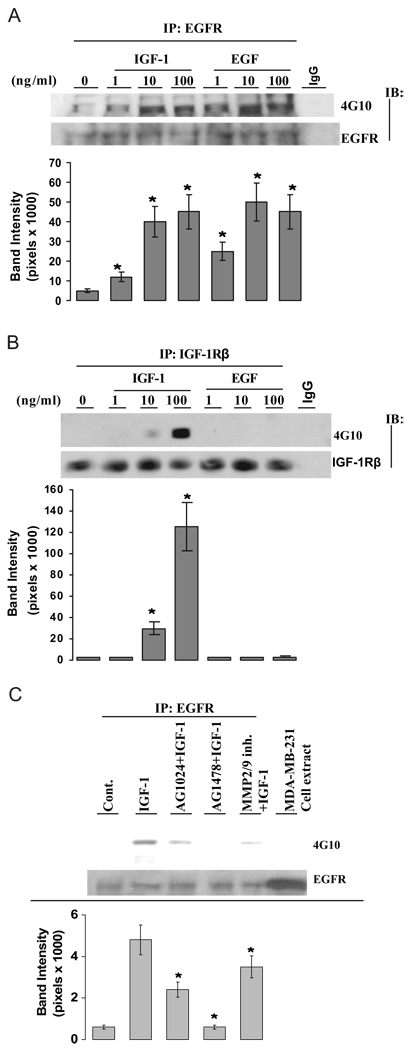
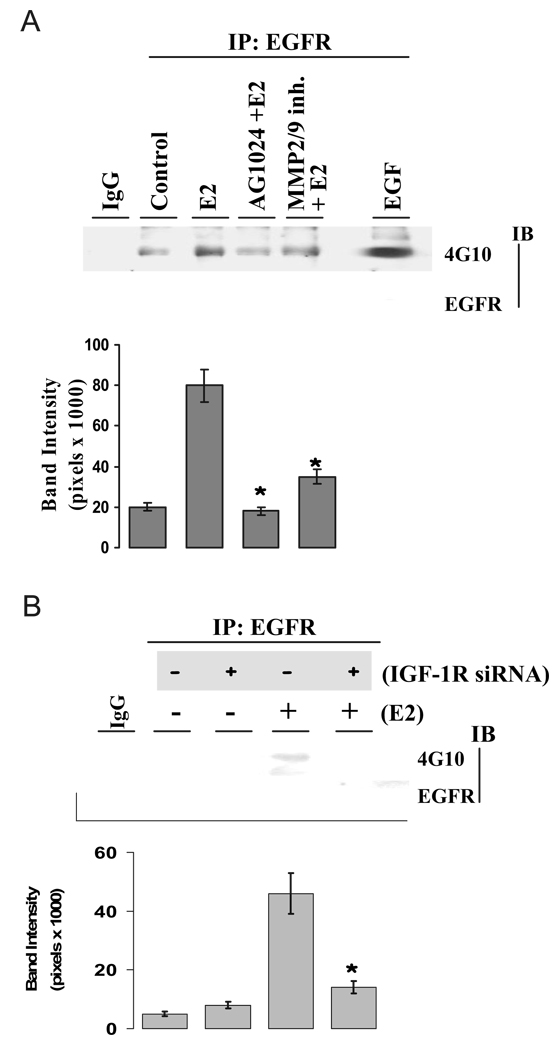
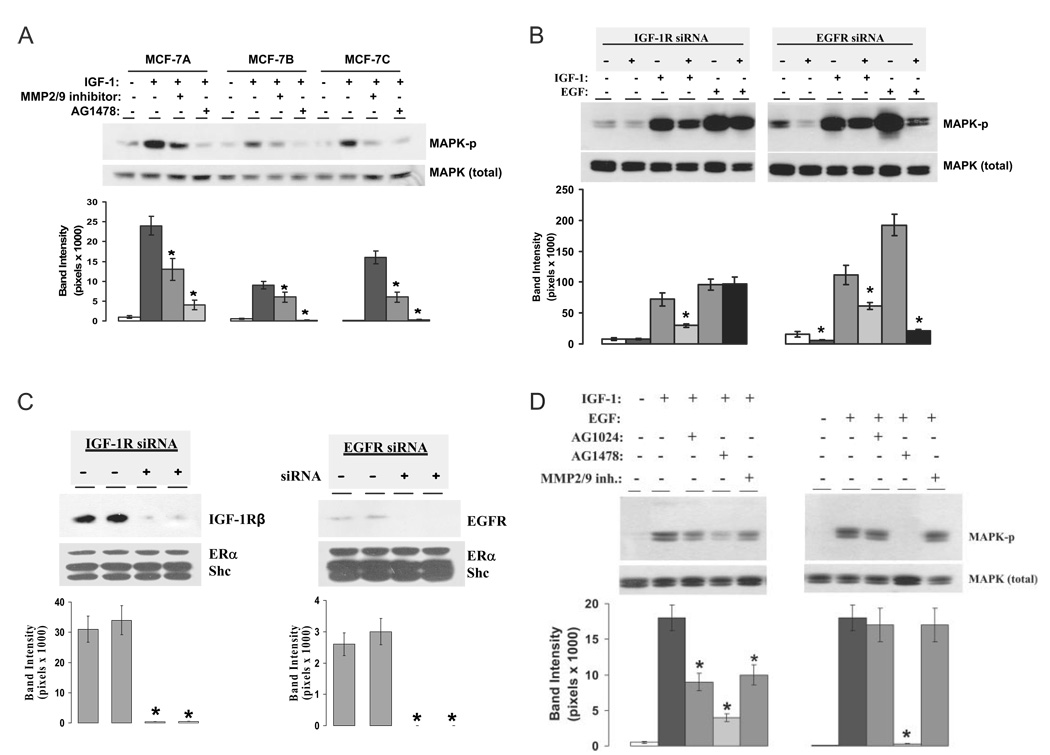
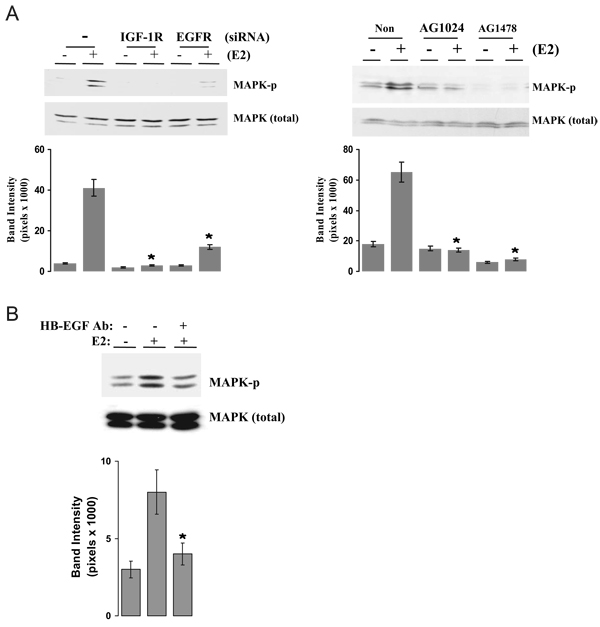
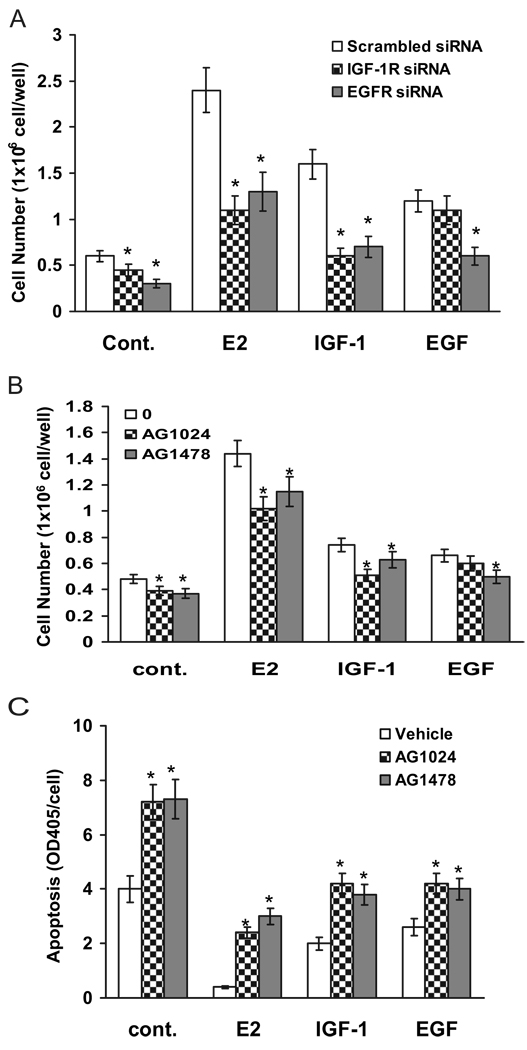
We present an integrated model of an extranuclear, estrogen receptor-alpha (ERalpha)-mediated, rapid MAPK activation pathway in breast cancer cells. In noncancer cells, IGF-I initiates a linear process involving activation of the IGF-I receptor (IGF-IR) and matrix metalloproteinases (MMP), release of heparin-binding epidermal growth factor (HB-EGF), and activation of EGF receptor (EGFR)-dependent MAPK. 17beta-Estradiol (E2) rapidly activates IGF-IR in breast cancer cells. We hypothesize that E2 induces a similar linear pathway involving IGF-IR, MMP, HB-EGF, EGFR, and MAPK. Using MCF-7 breast cancer cells, we for the first time demonstrated that a sequential activation of IGF-IR, MMP, and EGFR existed in E2 and IGF-I actions, which was supported by evidence that the selective inhibitors of IGF-IR and MMP or knockdown of IGF-IR all inhibited E2- or IGF-I-induced EGFR phosphorylation. Using the inhibitors and small inhibitory RNA strategies, we also demonstrated that the same sequential activation of the receptors occurred in E2-, IGF-I-, but not EGF-induced MAPK phosphorylation. Additionally, a HB-EGF neutralizing antibody significantly blocked E2-induced MAPK activation, further supporting our hypothesis. The biological effects of sequential activation of IGF-IR and EGFR on E2 stimulation of cell proliferation were also investigated. Knockdown or blockade of IGF-IR significantly inhibited E2- or IGF-I-stimulated but not EGF-induced cell growth. Knockdown or blockade of EGFR abrogated cell growth induced by E2, IGF-I, and EGF, indicating that EGFR is a downstream molecule of IGF-IR in E2 and IGF-I action. Together, our data support the novel view that E2 can activate a linear pathway involving the sequential activation of IGF-IR, MMP, HB-EGF, EGFR, and MAPK.
Figures
References
-
- Ali S, Coombes RC. Estrogen receptor α in human breast cancer: occurrence and significance. J Mammary Gland Biol Neoplasia. 2000;5:271–281. - PubMed
-
- Yee D, Lee AV. Crosstalk between the insulin-like growth factors and estrogens in breast cancer. J Mammary Gland Biol Neoplasia. 2000;5:107–115. - PubMed
-
- Cheskis BJ. Regulation of cell signalling cascades by steroid hormones. J Cell Biochem. 2004;93:20–27. - PubMed
-
- Alexaki VI, Charalampopoulos I, Kampa M, Vassalou H, Theodoropoulos P, Stathopoulos EN, Hatzoglou A, Gravanis A, Castanas E. Estrogen exerts neuroprotective effects via membrane estrogen receptors and rapid Akt/NOS activation. FASEB J. 2004;18:1594–1596. - PubMed
-
- Nicholson S, Wright C, Sainsbury JR, Halcrow P, Kelly P, Angus B, Farndon JR, Harris AL. Epidermal growth factor receptor (EGFr) as a marker for poor prognosis in node-negative breast cancer patients: neu and tamoxifen failure. J Steroid Biochem Mol Biol. 1990;37:811–814. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
