

Molecular dynamics simulations of the native and partially folded states of ubiquitin: influence of methanol cosolvent, pH, and temperature on the protein structure and dynamics
- PMID: 17525462
- PMCID: PMC2206653
- DOI: 10.1110/ps.062323407
Molecular dynamics simulations of the native and partially folded states of ubiquitin: influence of methanol cosolvent, pH, and temperature on the protein structure and dynamics
Abstract
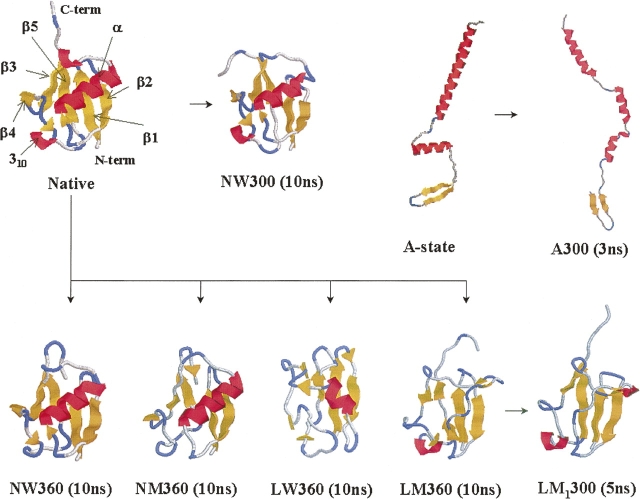
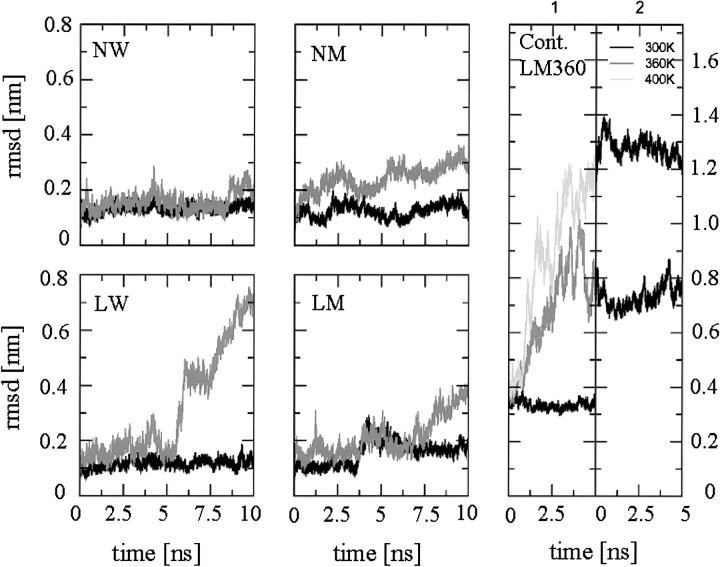
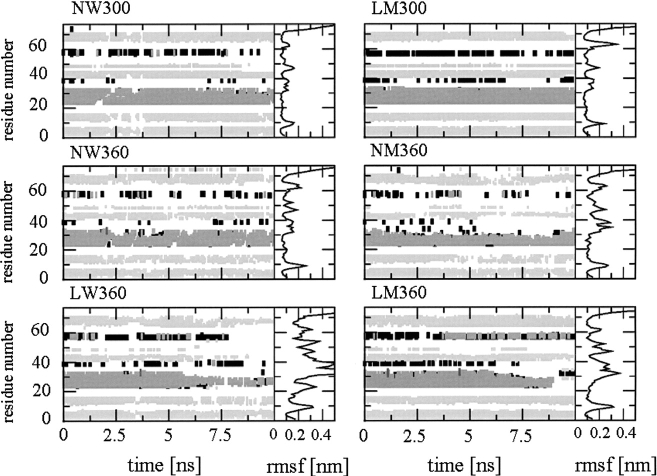
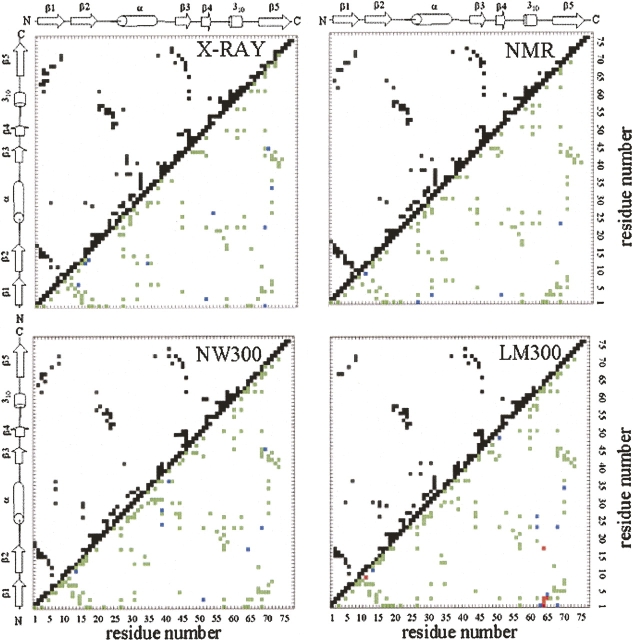
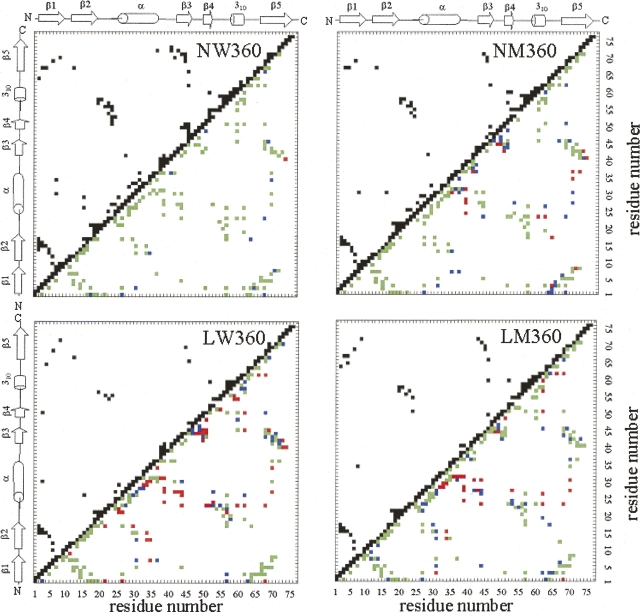
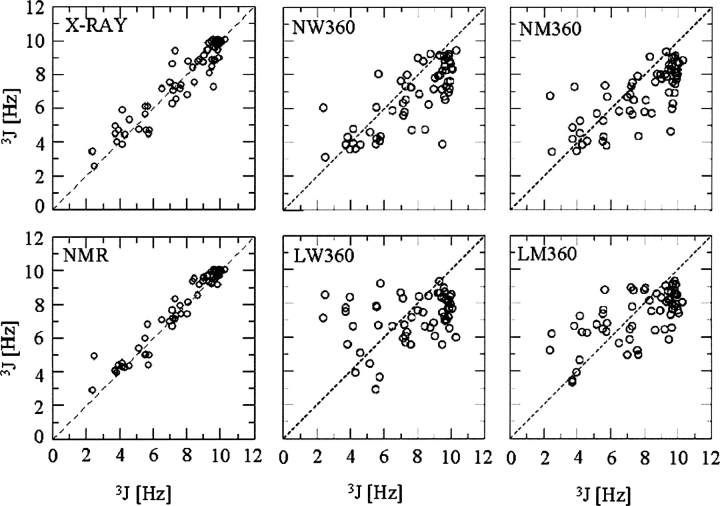
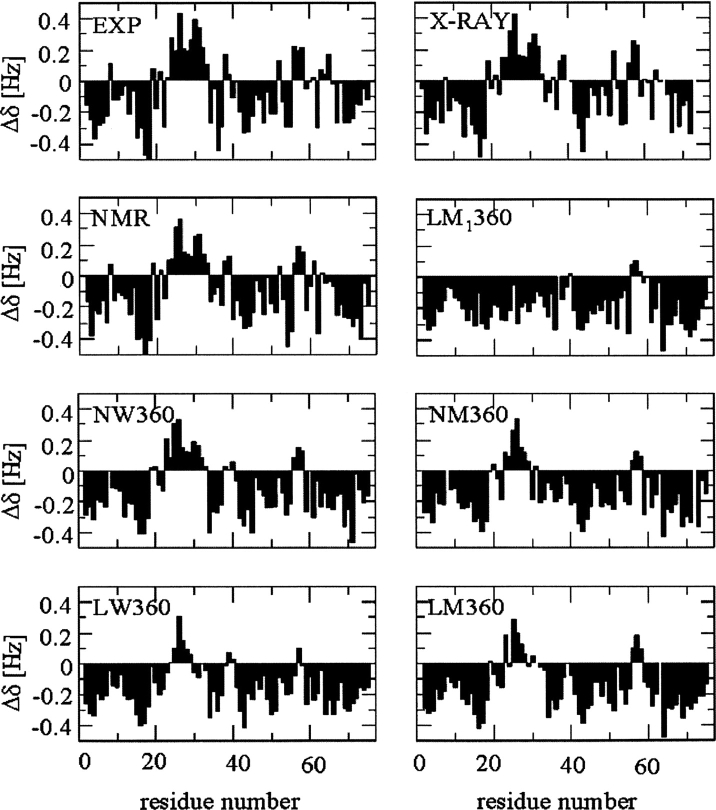
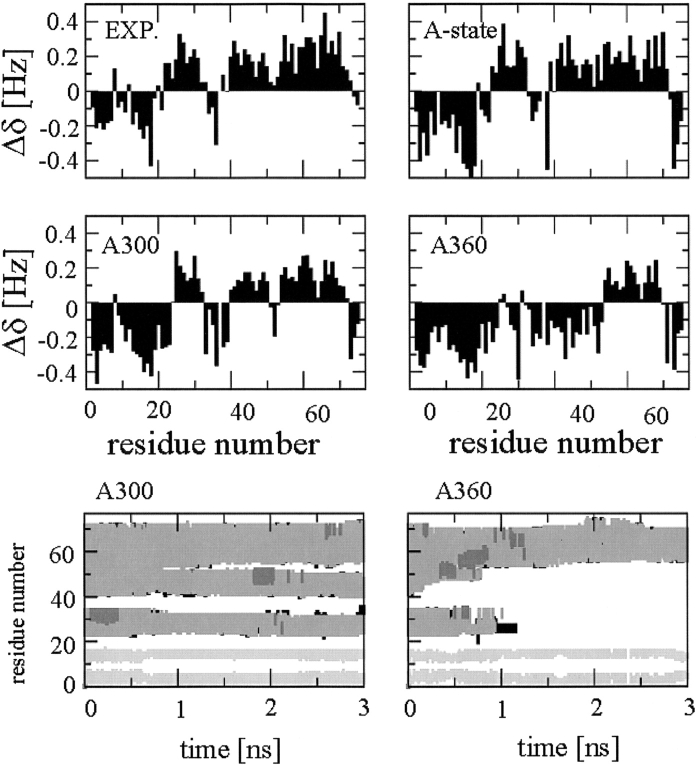
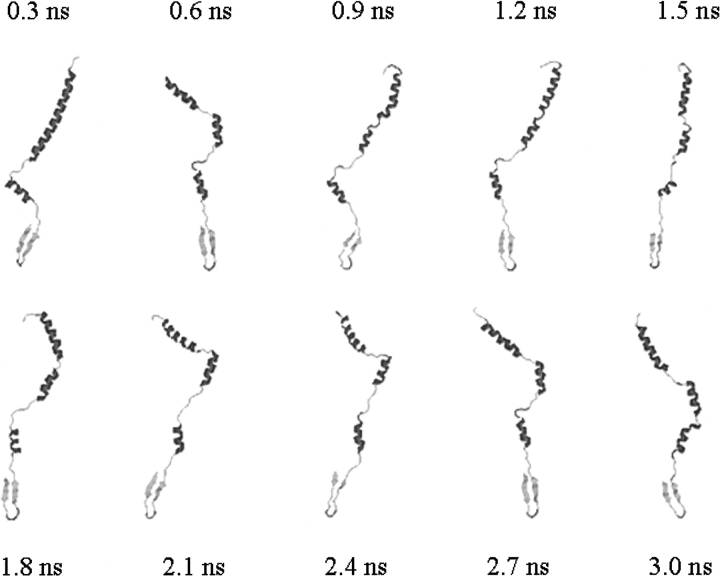
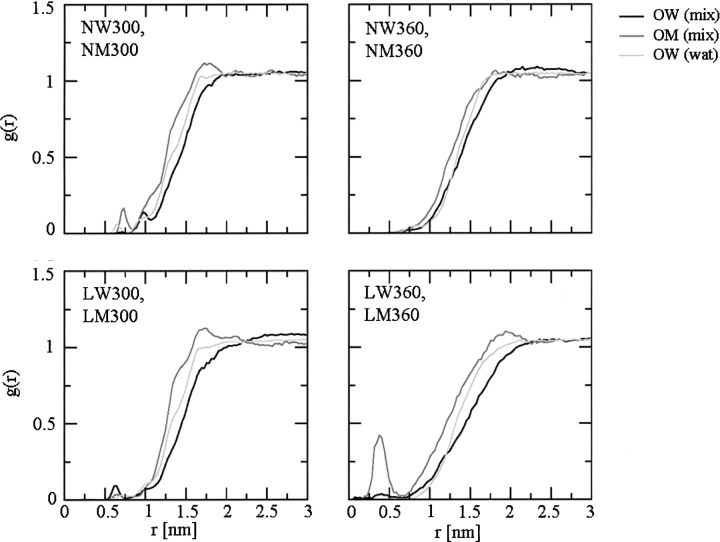
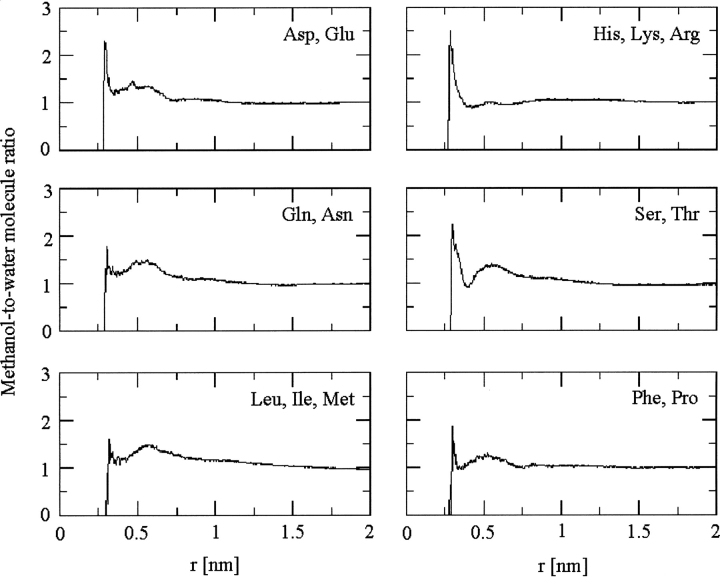
A series of explicit-solvent molecular dynamics simulations of the protein ubiquitin are reported, which investigate the effect of environmental factors (presence of methanol cosolvent in the aqueous solution, neutral or low pH value, room or elevated temperature) on the structure, stability, and dynamics of the protein. The simulations are initiated either from the native structure of the protein or from a model of a partially folded state (A-state) that is known to exist at low pH in methanol-water mixtures. The main results of the simulations are: (1) The ubiquitin native structure is remarkably stable at neutral pH in water; (2) the addition of the methanol cosolvent enhances the stability of the secondary structure but weakens tertiary interactions within the protein; (3) this influence of methanol on the protein structure is enhanced at low pH, while the effect of lowering the pH in pure water is limited; and (4) the A-state of ubiquitin can be described as a set of relatively rigid secondary structure elements (a native-like beta-sheet and native-like alpha-helix plus two nonnative alpha-helices) connected by flexible linkers.
Figures

References
-
- Alexandrescu A.T., Ng, Y.L., and Dobson, C.M. 1994. Characterization of a trifluoroethanol-induced partially folded state of α-lactalbumin. J. Mol. Biol. 235: 587–599. - PubMed
-
- Alonso D.O. and Daggett, V. 1995. Molecular dynamics simulations of protein unfolding and limited refolding: Characterization of partially unfolded states of ubiquitin in 60% methanol and in water. J. Mol. Biol. 247: 501–520. - PubMed
-
- Arai M. and Kuwajima, K. 2000. Role of the molten globule state in protein folding. Adv. Protein Chem. 53: 209–282. - PubMed
-
- Barker J.A. and Watts, R.O. 1973. Monte-Carlo studies of the dielectric properties of water-like models. Mol. Phys. 26: 789–792.
-
- Berendsen H.J.C., Postma, J.P.M., van Gunsteren, W.F., and Hermans, J. 1981. Interaction models for water in relation to protein hydration. Jerusalem Symposia on Quantum Chemistry and Biochemistry, Vol. 14, pp. 331–342. Reidel, Dordrecht, Holland.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
