

Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones
- PMID: 17525798
- PMCID: PMC1868788
- DOI: 10.1172/JCI31561
Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones
Abstract
PPAR gamma is required for fat cell development and is the molecular target of antidiabetic thiazolidinediones (TZDs), which exert insulin-sensitizing effects in adipose tissue, skeletal muscle, and liver. Unexpectedly, we found that inactivation of PPAR gamma in macrophages results in the development of significant glucose intolerance plus skeletal muscle and hepatic insulin resistance in lean mice fed a normal diet. This phenotype was associated with increased expression of inflammatory markers and impaired insulin signaling in adipose tissue, muscle, and liver. PPAR gamma-deficient macrophages secreted elevated levels of factors that impair insulin responsiveness in muscle cells in a manner that was enhanced by exposure to FFAs. Consistent with this, the relative degree of insulin resistance became more severe in mice lacking macrophage PPAR gamma following high-fat feeding, and these mice were only partially responsive to TZD treatment. These findings reveal an essential role of PPAR gamma in macrophages for the maintenance of whole-body insulin action and in mediating the antidiabetic actions of TZDs.
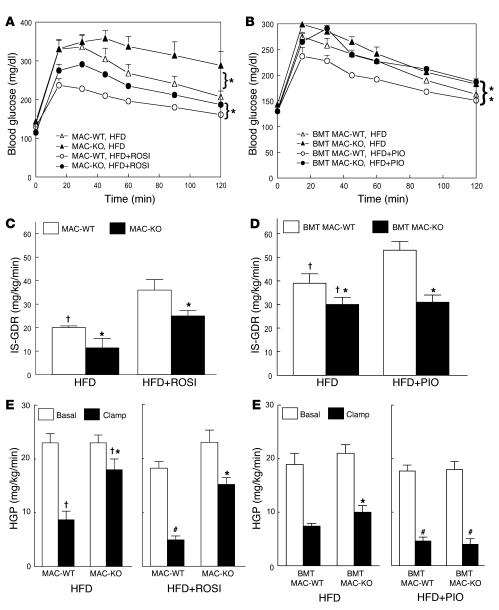
Figures
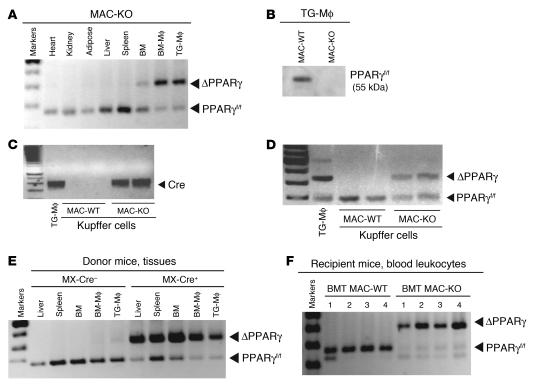
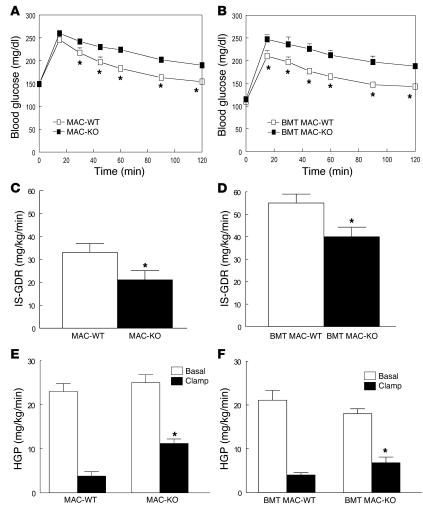
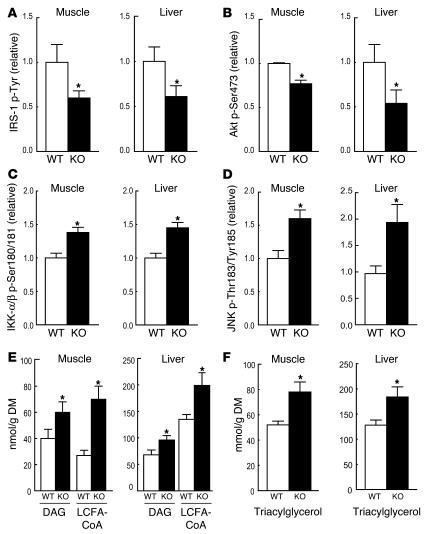

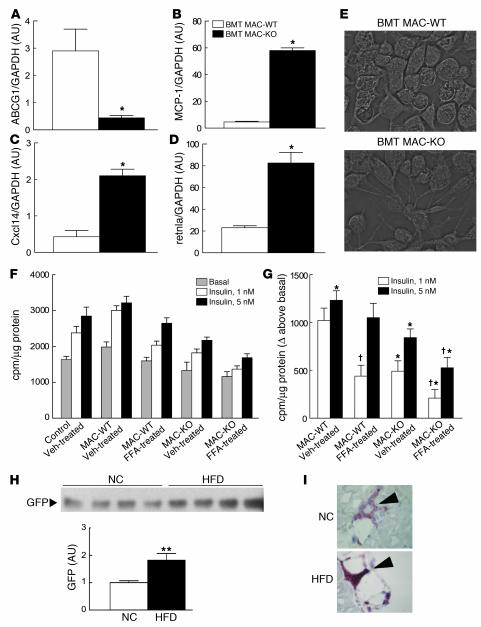
References
-
- DeFronzo R.A., Bonadonna R.C., Ferrannini E. Pathogenesis of NIDDM. A balanced overview. Diabetes Care. 1992;15:318–368. - PubMed
-
- Reaven G.M. Role of insulin resistance in human disease (syndrome X): an expanded definition. Annu. Rev. Med. 1993;44:121–131. - PubMed
-
- Miranda P.J., DeFronzo R.A., Califf R.M., Guyton J.R. Metabolic syndrome: definition, pathophysiology, and mechanisms. Am. Heart J. 2005;149:33–45. - PubMed
-
- Arkan M.C., et al. IKK-β links inflammation to obesity-induced insulin resistance. Nat. Med. 2005;11:191–198. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- GM069338/GM/NIGMS NIH HHS/United States
- DK60484/DK/NIDDK NIH HHS/United States
- P01 DK074868/DK/NIDDK NIH HHS/United States
- DK33651/DK/NIDDK NIH HHS/United States
- U54 GM069338/GM/NIGMS NIH HHS/United States
- R01 DK033651/DK/NIDDK NIH HHS/United States
- K01 DK060484/DK/NIDDK NIH HHS/United States
- P30 DK063491/DK/NIDDK NIH HHS/United States
- HL56989/HL/NHLBI NIH HHS/United States
- P50 HL056989/HL/NHLBI NIH HHS/United States
- DK73227/DK/NIDDK NIH HHS/United States
- DK063491/DK/NIDDK NIH HHS/United States
- R21 DK073227/DK/NIDDK NIH HHS/United States
- DK074868/DK/NIDDK NIH HHS/United States
- R37 DK033651/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
