

Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern
- PMID: 17534432
- PMCID: PMC1868965
- DOI: 10.1371/journal.pone.0000482
Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern
Abstract
Background: Idiopathic pulmonary fibrosis (IPF) is characterized by the insidious onset of dyspnea or cough. However, a subset of patients has a short duration of symptoms with rapid progression to end-stage disease. In this study, we evaluated clinical and molecular features of "rapid" and "slow" progressors with IPF.
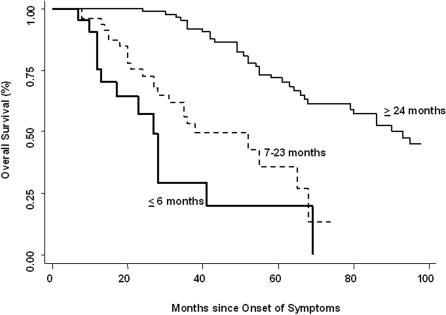
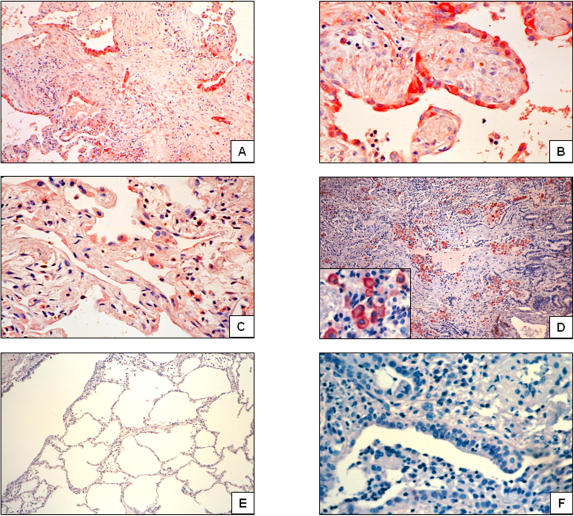
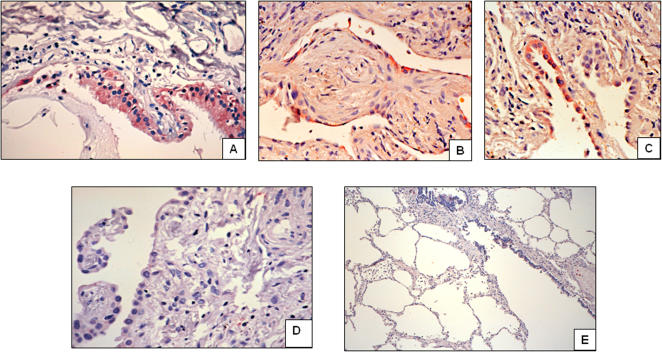
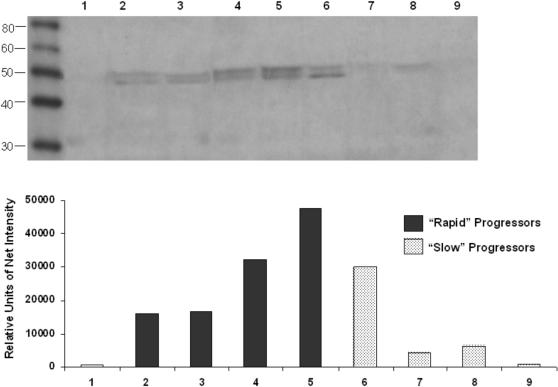
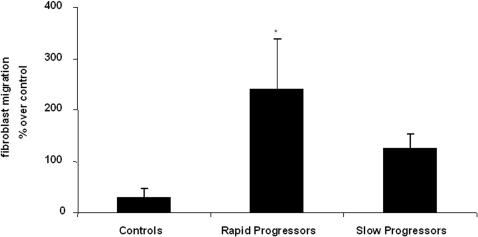
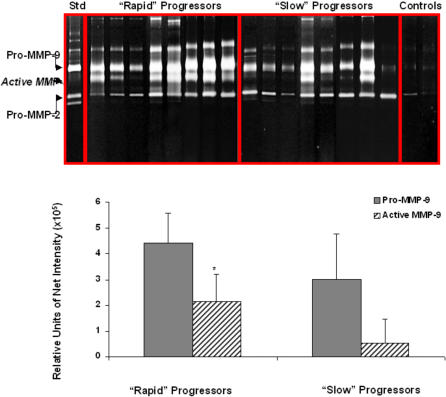
Methods and findings: 26 patients with <6 months of symptoms before first presentation [rapid progressors] and 88 patients with >24 months of symptoms [slow progressors] were studied. Survival was analyzed by the Kaplan-Meyer method and proportional hazard's model. Lung microarrays and tissue proteins were measured in a subset of patients. No differences were found in age, physiologic impairment and bronchoalveolar lavage (BAL) cellular profile. There were more males (OR = 6.5; CI:1.4-29.5; p = 0.006) and smokers (OR = 3.04; CI:1.1-8.3; p = 0.04) in the rapid progressors group. Survival from the beginning of symptoms was significantly reduced in rapid progressors (HR = 9.0; CI:4.48-18.3; p<0.0001) and there was a tendency for decreased survival from the time of diagnosis (HR = 1.5; CI:0.81-2.87; p = 0.18). We identified 437 differentially expressed genes. Lungs of rapid progressors overexpressed genes involved in morphogenesis, oxidative stress, migration/proliferation, and genes from fibroblasts/smooth muscle cells. Upregulation of two of these genes, adenosine-2B receptor and prominin-1/CD133, was validated by immunohistochemistry and were expressed by alveolar epithelial cells. BAL from rapid progressors showed a >2-fold increase of active matrix metalloproteinase-9, and induced a higher fibroblast migration compared with slow progressors and controls [238+/-98% versus 123+/-29% (p<0.05) and 30+/-17% (p<0.01)].
Conclusions/significance: A subgroup of IPF patients, predominantly smoking males, display an accelerated clinical course and have a gene expression pattern that is different from those with slower progression and longer survival. These findings highlight the variability in the progression of IPF, and may explain, in part, the difficulty in obtaining significant and reproducible results in studies of therapeutic interventions in patients with IPF.
Conflict of interest statement
Figures

References
-
- Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345:517–525. - PubMed
-
- Collard HR, Ryu JH, Douglas WW, Schwarz MI, Curran-Everett D, et al. Combined corticosteroid and cyclophosphamide therapy does not alter survival in idiopathic pulmonary fibrosis. Chest. 2004;125:2169–2174. - PubMed
-
- Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. - PubMed
-
- McCormack FX, King TE, Jr, Bucher BL, Nielsen L, Mason RJ. Surfactant protein A predicts survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1995;152:751–759. - PubMed
-
- Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142:963–967. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
