

Inhibition of ubiquitin-mediated degradation of MOAP-1 by apoptotic stimuli promotes Bax function in mitochondria
- PMID: 17535899
- PMCID: PMC1877986
- DOI: 10.1073/pnas.0700007104
Inhibition of ubiquitin-mediated degradation of MOAP-1 by apoptotic stimuli promotes Bax function in mitochondria
Abstract
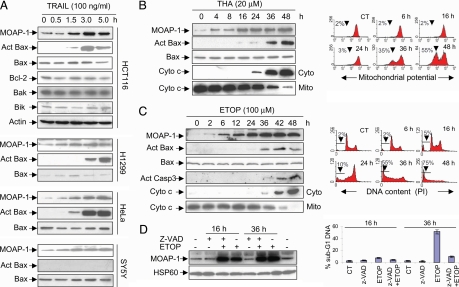
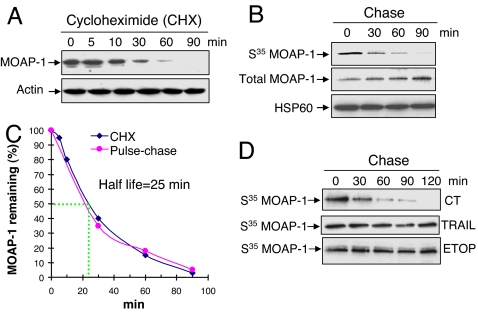
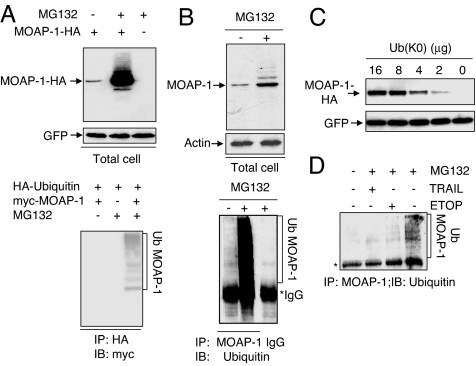
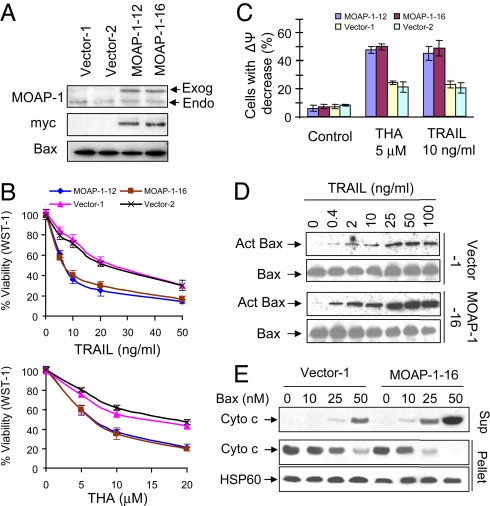
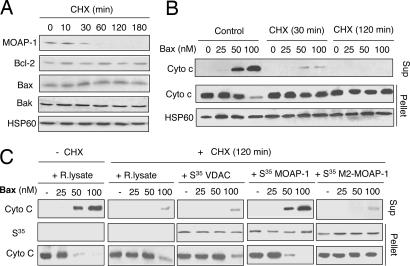
The multidomain proapoptotic protein Bax of the Bcl-2 family is a central regulator for controlling the release of apoptogenic factors from mitochondria. Recent evidence suggests that the Bax-associating protein MOAP-1 may act as an effector for promoting Bax function in mitochondria. Here, we report that MOAP-1 protein is rapidly up-regulated by multiple apoptotic stimuli in mammalian cells. MOAP-1 is a short-lived protein (t(1/2) approximately 25 min) that is constitutively degraded by the ubiquitin-proteasome system. Induction of MOAP-1 by apoptotic stimuli ensues through inhibition of its polyubiquitination process. Elevation of MOAP-1 levels sensitizes cells to apoptotic stimuli and promotes recombinant Bax-mediated cytochrome c release from isolated mitochondria. Mitochondria depleted of short-lived proteins by cycloheximide (CHX) become resistant to Bax-mediated cytochrome c release. Remarkably, incubation of these mitochondria with in vitro-translated MOAP-1 effectively restores the cytochrome c releasing effect of recombinant Bax. We propose that apoptotic stimuli can facilitate the proapoptotic function of Bax in mitochondria through stabilization of MOAP-1.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
