

Evidence that mono-ADP-ribosylation of CtBP1/BARS regulates lipid storage
- PMID: 17538025
- PMCID: PMC1949384
- DOI: 10.1091/mbc.e06-09-0869
Evidence that mono-ADP-ribosylation of CtBP1/BARS regulates lipid storage
Abstract
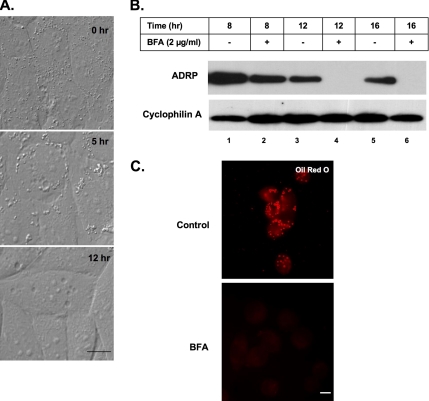
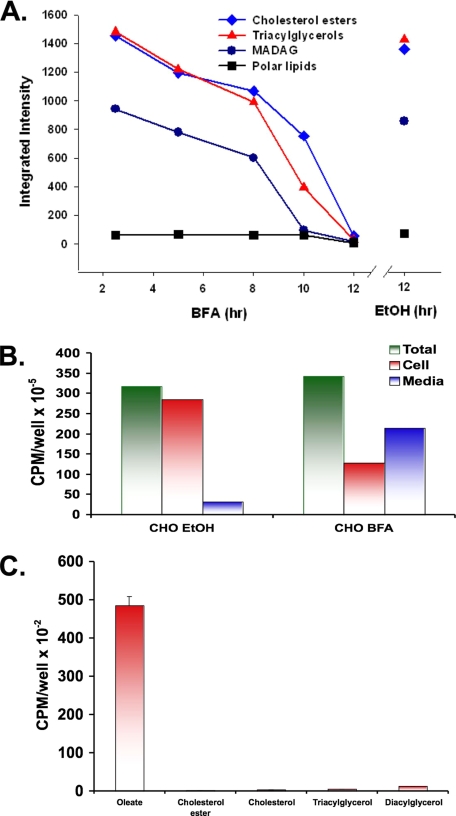
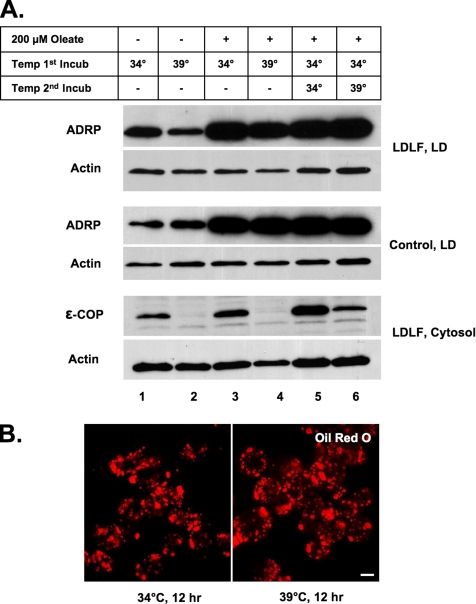
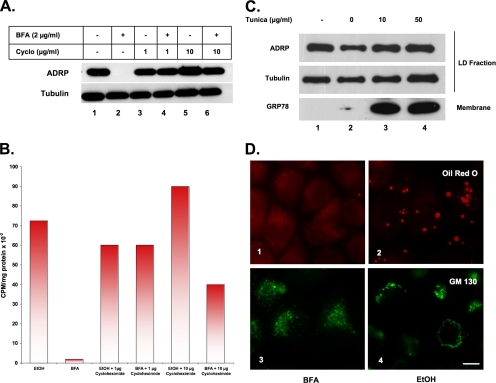
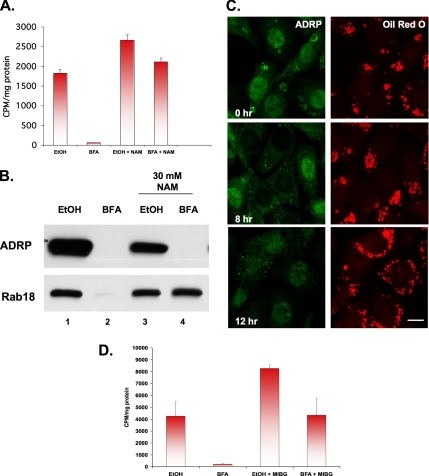
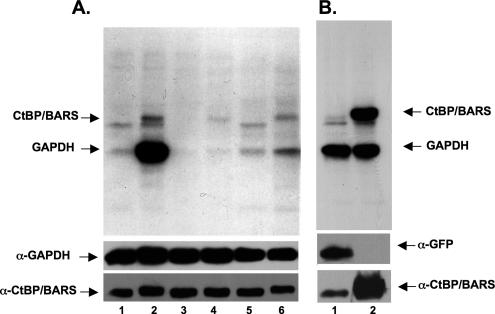
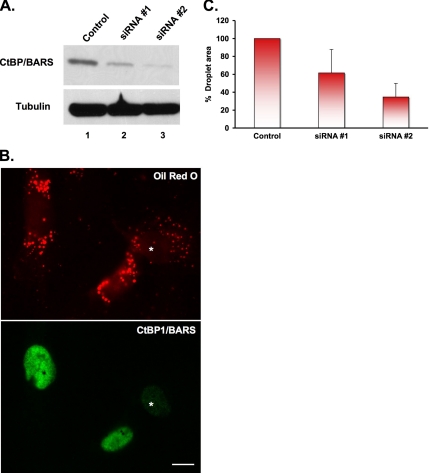
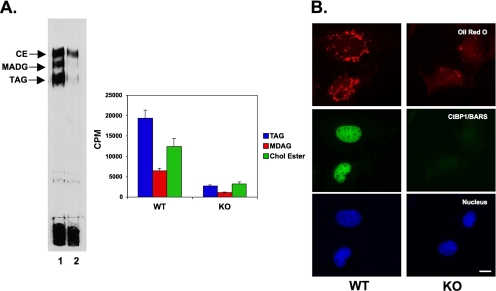
Mono-ADP-ribosylation is emerging as an important posttranslational modification that modulates a variety of cell signaling pathways. Here, we present evidence that mono-ADP-ribosylation of the transcriptional corepressor C terminal binding protein, brefeldin A (BFA)-induced ADP-ribosylated substrate (CtBP1/BARS) regulates neutral lipid storage in droplets that are surrounded by a monolayer of phospholipid and associated proteins. CtBP1/BARS is an NAD-binding protein that becomes ribosylated when cells are exposed to BFA. Both endogenous lipid droplets and droplets enlarged by oleate treatment are lost after 12-h exposure to BFA. Lipid loss requires new protein synthesis, and it is blocked by multiple ribosylation inhibitors, but it is not stimulated by disruption of the Golgi apparatus or the endoplasmic reticulum unfolded protein response. Small interfering RNA knockdown of CtBP1/BARS mimics the effect of BFA, and mouse embryonic fibroblasts derived from embryos that are deficient in CtBP1/BARS seem to be defective in lipid accumulation. We conclude that mono-ADP-ribosylation of CtBP1/BARS inactivates its repressor function, which leads to the activation of genes that regulate neutral lipid storage.
Figures
References
-
- Agarwal A. K., Auchus R. J. Minireview: cellular redox state regulates hydroxysteroid dehydrogenase activity and intracellular hormone potency. Endocrinology. 2005;146:2531–2538. - PubMed
-
- Bartz R., Li W. H., Venables B., Zehmer J. K., Roth M. R., Welti R., Anderson R. G., Liu P., Chapman K. D. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J. Lipid Res. 2007;48:837–847. - PubMed
-
- Bligh E. G., Dyer W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. - PubMed
-
- Brasaemle D. L., Dolios G., Shapiro L., Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3–L1 adipocytes. J. Biol. Chem. 2004;279:46835–46842. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
