

Characterization and modeling of the Haemophilus influenzae core and supragenomes based on the complete genomic sequences of Rd and 12 clinical nontypeable strains
- PMID: 17550610
- PMCID: PMC2394751
- DOI: 10.1186/gb-2007-8-6-r103
Characterization and modeling of the Haemophilus influenzae core and supragenomes based on the complete genomic sequences of Rd and 12 clinical nontypeable strains
Abstract
Background: The distributed genome hypothesis (DGH) posits that chronic bacterial pathogens utilize polyclonal infection and reassortment of genic characters to ensure persistence in the face of adaptive host defenses. Studies based on random sequencing of multiple strain libraries suggested that free-living bacterial species possess a supragenome that is much larger than the genome of any single bacterium.
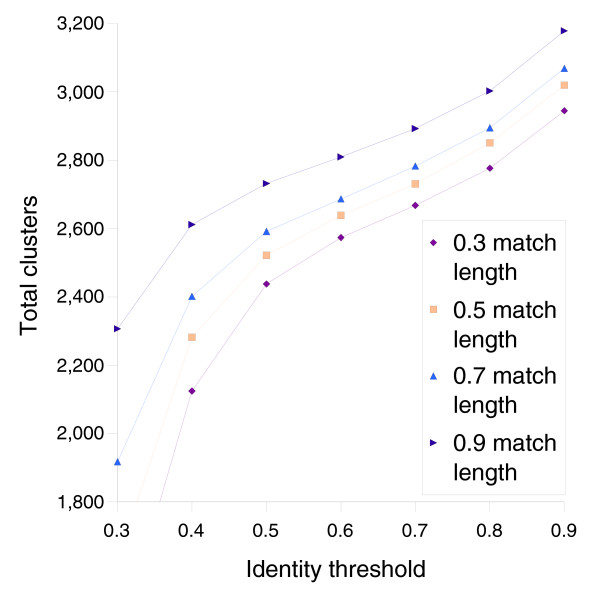
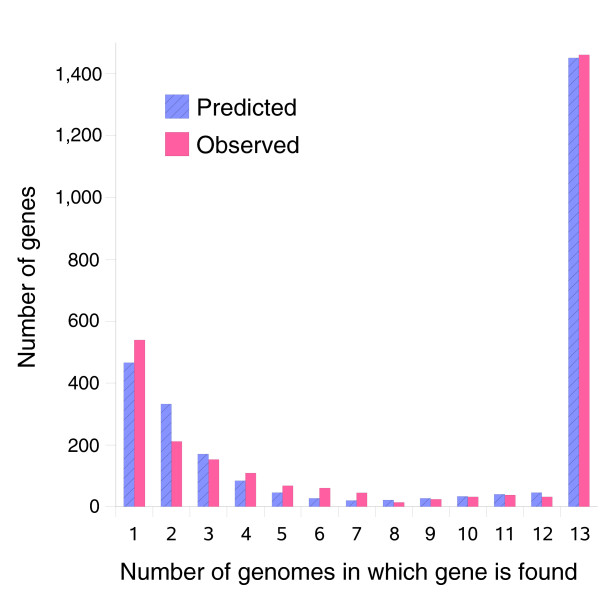
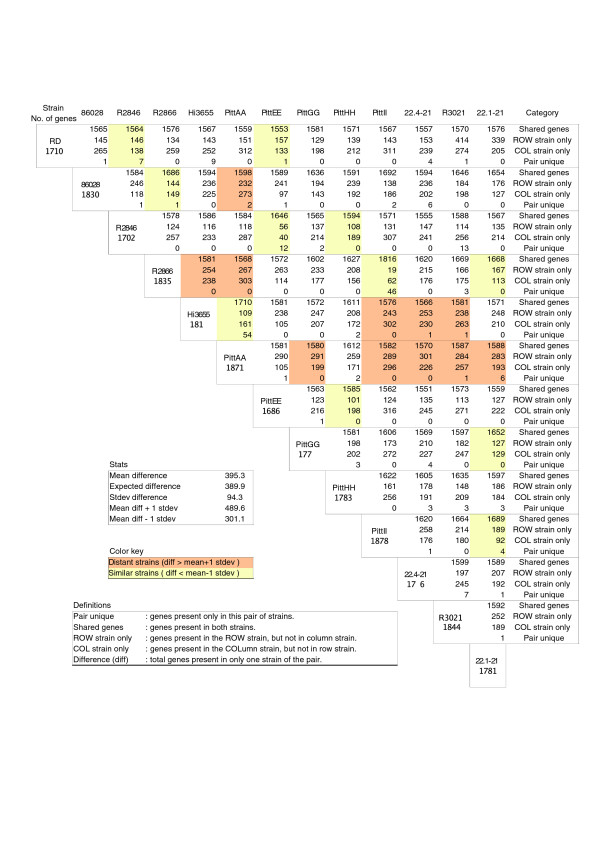
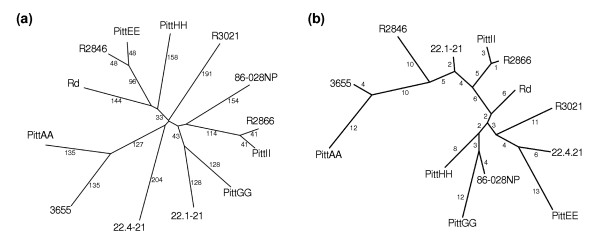
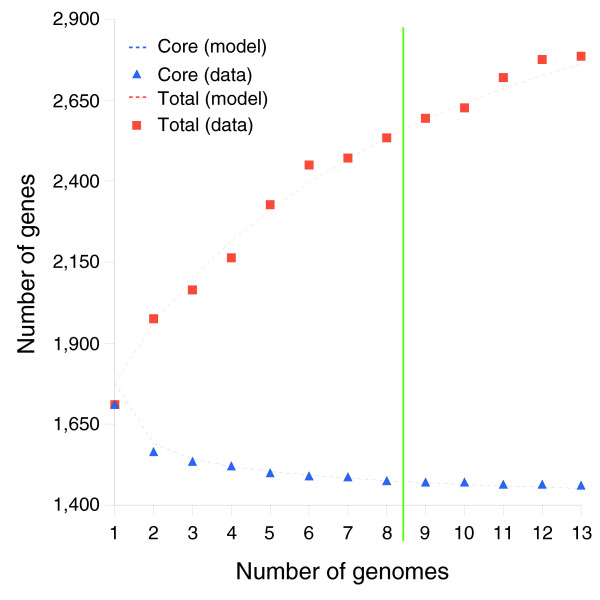
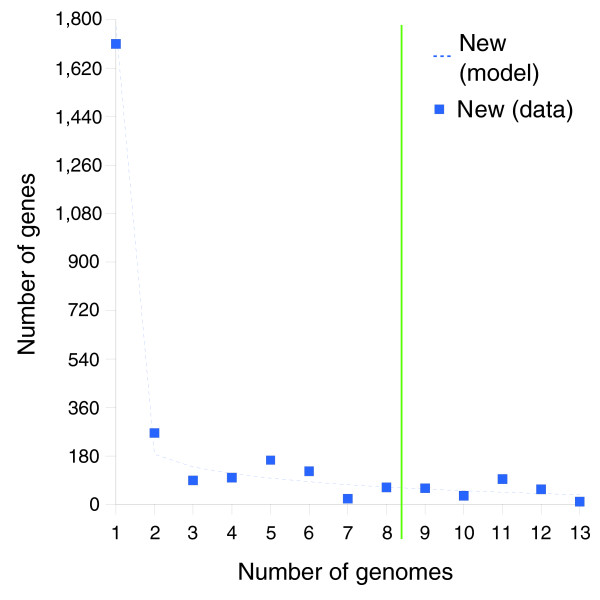
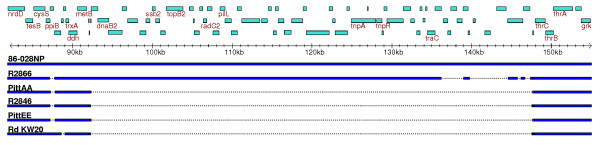
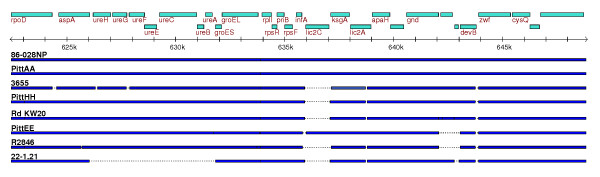
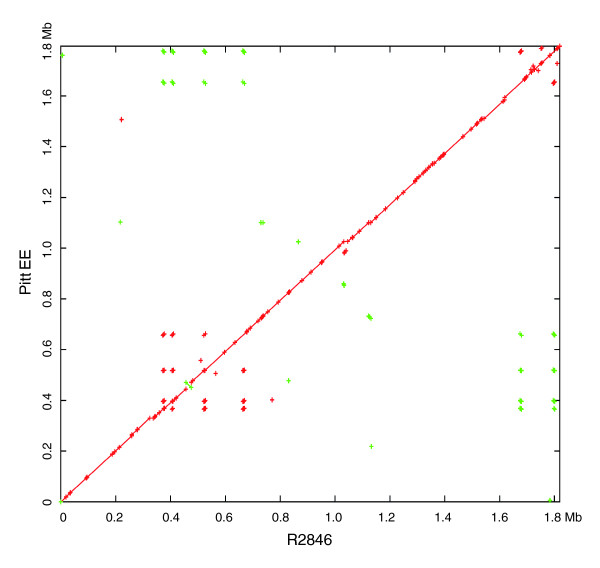
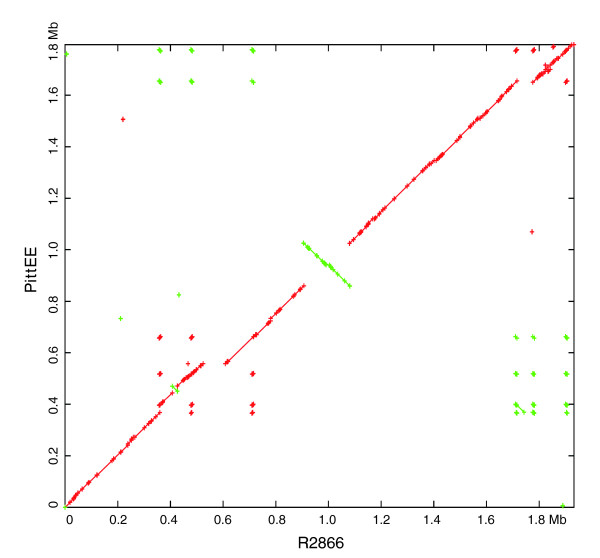
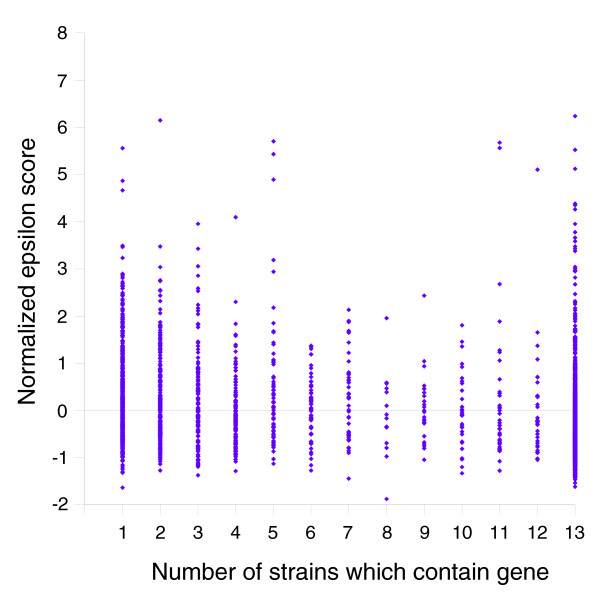
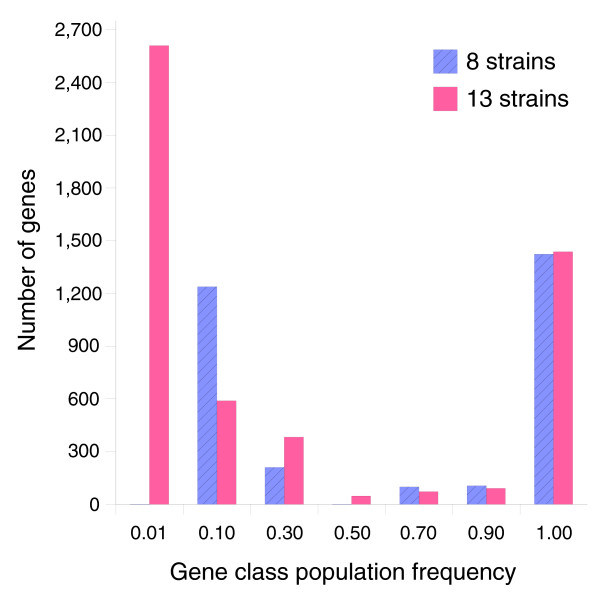
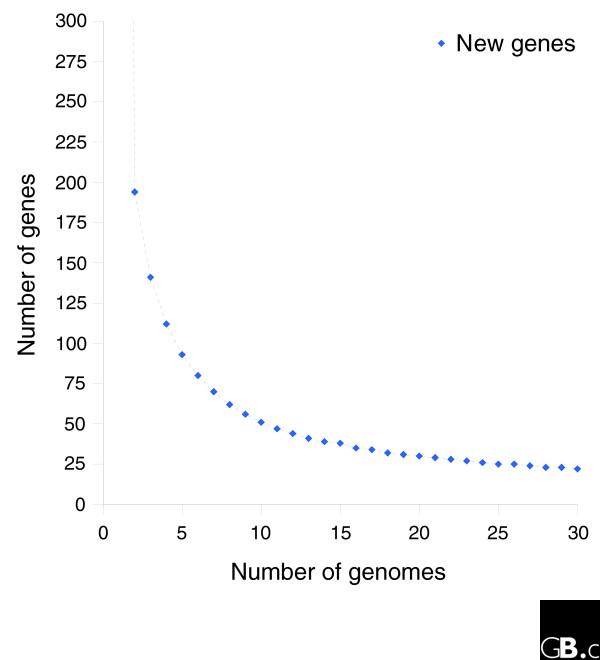
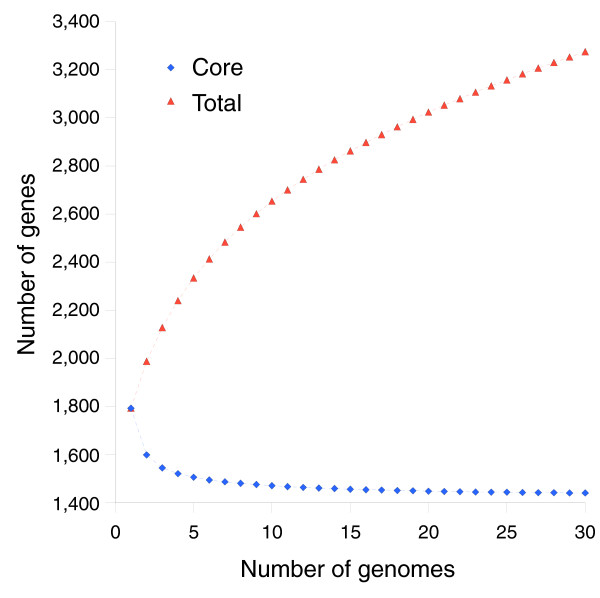
Results: We derived high depth genomic coverage of nine nontypeable Haemophilus influenzae (NTHi) clinical isolates, bringing to 13 the number of sequenced NTHi genomes. Clustering identified 2,786 genes, of which 1,461 were common to all strains, with each of the remaining 1,328 found in a subset of strains; the number of clusters ranged from 1,686 to 1,878 per strain. Genic differences of between 96 and 585 were identified per strain pair. Comparisons of each of the NTHi strains with the Rd strain revealed between 107 and 158 insertions and 100 and 213 deletions per genome. The mean insertion and deletion sizes were 1,356 and 1,020 base-pairs, respectively, with mean maximum insertions and deletions of 26,977 and 37,299 base-pairs. This relatively large number of small rearrangements among strains is in keeping with what is known about the transformation mechanisms in this naturally competent pathogen.
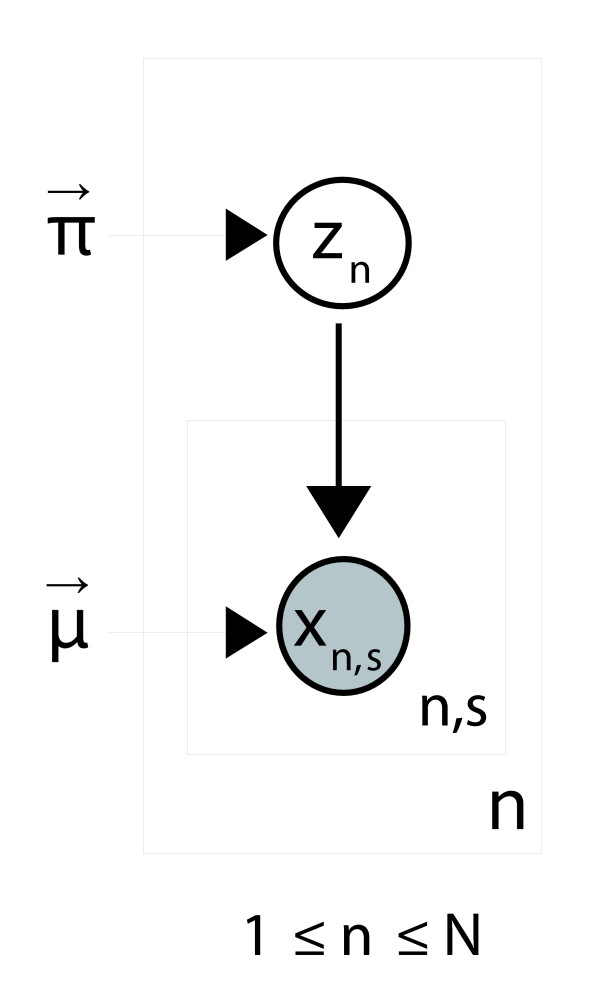
Conclusion: A finite supragenome model was developed to explain the distribution of genes among strains. The model predicts that the NTHi supragenome contains between 4,425 and 6,052 genes with most uncertainty regarding the number of rare genes, those that have a frequency of <0.1 among strains; collectively, these results support the DGH.
Figures

References
-
- Ehrlich GD, Veeh R, Wang X, Costerton JW, Hayes JD, Hu FZ, Daigle BJ, Ehrlich MD. Mucosal biofilm formation on middle-ear mucosa in the chinchilla model of otitis media. JAMA. 2002;287:1710–1715. - PubMed
-
- Post JC, Preston RA, Aul JJ, Larkins-Pettigrew M, Rydquist-White J, Anderson KW, Wadowsky RM, Reagan DR, Walker ES, Kingsley LA, et al. Molecular analysis of bacterial pathogens in otitis media with effusion. JAMA. 1995;273:1598–1604. - PubMed
-
- Murphy TF, Sethi S, Klingman KL, Brueggemann AB, Doern GV. Simultaneous respiratory tract colonization by multiple strains of nontypeable Haemophilus influenzae in chronic obstructive pulmonary disease: implications for antibiotic therapy. J Infect Dis. 1999;180:404–409. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
