

Systematic prediction and validation of breakpoints associated with copy-number variants in the human genome
- PMID: 17551006
- PMCID: PMC1891248
- DOI: 10.1073/pnas.0703834104
Systematic prediction and validation of breakpoints associated with copy-number variants in the human genome
Abstract
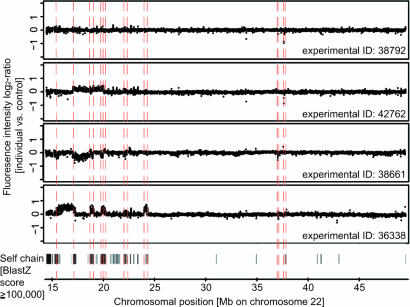
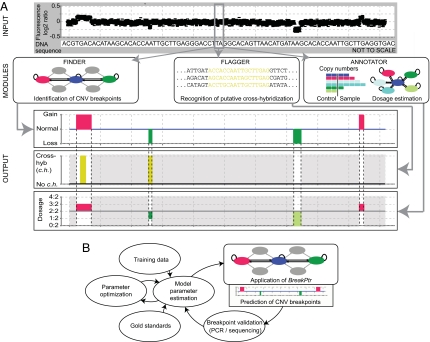
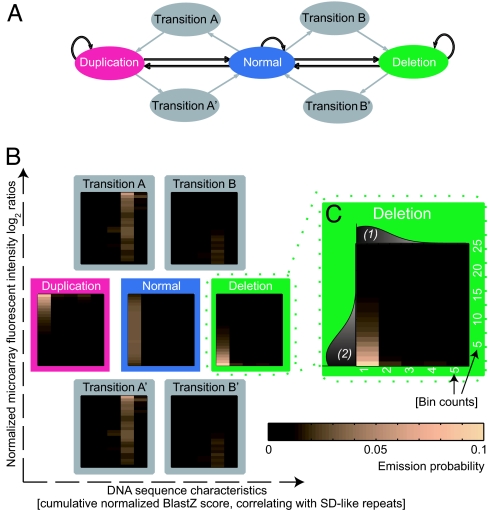
Copy-number variants (CNVs) are an abundant form of genetic variation in humans. However, approaches for determining exact CNV breakpoint sequences (physical deletion or duplication boundaries) across individuals, crucial for associating genotype to phenotype, have been lacking so far, and the vast majority of CNVs have been reported with approximate genomic coordinates only. Here, we report an approach, called BreakPtr, for fine-mapping CNVs (available from http://breakptr.gersteinlab.org). We statistically integrate both sequence characteristics and data from high-resolution comparative genome hybridization experiments in a discrete-valued, bivariate hidden Markov model. Incorporation of nucleotide-sequence information allows us to take into account the fact that recently duplicated sequences (e.g., segmental duplications) often coincide with breakpoints. In anticipation of an upcoming increase in CNV data, we developed an iterative, "active" approach to initially scoring with a preliminary model, performing targeted validations, retraining the model, and then rescoring, and a flexible parameterization system that intuitively collapses from a full model of 2,503 parameters to a core one of only 10. Using our approach, we accurately mapped >400 breakpoints on chromosome 22 and a region of chromosome 11, refining the boundaries of many previously approximately mapped CNVs. Four predicted breakpoints flanked known disease-associated deletions. We validated an additional four predicted CNV breakpoints by sequencing. Overall, our results suggest a predictive resolution of approximately 300 bp. This level of resolution enables more precise correlations between CNVs and across individuals than previously possible, allowing the study of CNV population frequencies. Further, it enabled us to demonstrate a clear Mendelian pattern of inheritance for one of the CNVs.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Analysis of chromosome breakpoints in neuroblastoma at sub-kilobase resolution using fine-tiling oligonucleotide array CGH.Genes Chromosomes Cancer. 2005 Nov;44(3):305-19. doi: 10.1002/gcc.20243. Genes Chromosomes Cancer. 2005. PMID: 16075461
-
High-resolution mapping of DNA copy alterations in human chromosome 22 using high-density tiling oligonucleotide arrays.Proc Natl Acad Sci U S A. 2006 Mar 21;103(12):4534-9. doi: 10.1073/pnas.0511340103. Epub 2006 Mar 14. Proc Natl Acad Sci U S A. 2006. PMID: 16537408 Free PMC article.
-
Molecular characterization of ring chromosome 18 by low-coverage next generation sequencing.BMC Med Genet. 2015 Jul 30;16:57. doi: 10.1186/s12881-015-0206-x. BMC Med Genet. 2015. PMID: 26224010 Free PMC article.
-
Origins and breakpoint analyses of copy number variations: up close and personal.Cytogenet Genome Res. 2011;135(3-4):271-6. doi: 10.1159/000330267. Epub 2011 Aug 12. Cytogenet Genome Res. 2011. PMID: 21846967 Review.
-
Methods and strategies for analyzing copy number variation using DNA microarrays.Nat Genet. 2007 Jul;39(7 Suppl):S16-21. doi: 10.1038/ng2028. Nat Genet. 2007. PMID: 17597776 Free PMC article. Review.
Cited by
-
Copy number variation detection and genotyping from exome sequence data.Genome Res. 2012 Aug;22(8):1525-32. doi: 10.1101/gr.138115.112. Epub 2012 May 14. Genome Res. 2012. PMID: 22585873 Free PMC article.
-
Germline DNA copy number variation in familial and early-onset breast cancer.Breast Cancer Res. 2012 Feb 7;14(1):R24. doi: 10.1186/bcr3109. Breast Cancer Res. 2012. PMID: 22314128 Free PMC article.
-
A method for calling copy number polymorphism using haplotypes.Front Genet. 2013 Sep 23;4:165. doi: 10.3389/fgene.2013.00165. eCollection 2013. Front Genet. 2013. PMID: 24069028 Free PMC article.
-
INCT in oncogenetics focusing on hereditary breast-colorectal carcinoma syndrome.BMC Proc. 2013 Apr 4;7 Suppl 2(Suppl 2):K19. doi: 10.1186/1753-6561-7-S2-K19. eCollection 2013. BMC Proc. 2013. PMID: 24764476 Free PMC article. No abstract available.
-
Evidence of convergent evolution in humans and macaques supports an adaptive role for copy number variation of the β-defensin-2 gene.Genome Biol Evol. 2014 Oct 27;6(11):3025-38. doi: 10.1093/gbe/evu236. Genome Biol Evol. 2014. PMID: 25349268 Free PMC article.
References
-
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, et al. Science. 2004;305:525–528. - PubMed
-
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Nat Genet. 2004;36:949–951. - PubMed
-
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. Nat Genet. 2005;37:727–732. - PubMed
-
- Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, et al. Science. 2005;307:1434–1440. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
