

Glycogen storage diseases: new perspectives
- PMID: 17552001
- PMCID: PMC4146814
- DOI: 10.3748/wjg.v13.i18.2541
Glycogen storage diseases: new perspectives
Abstract
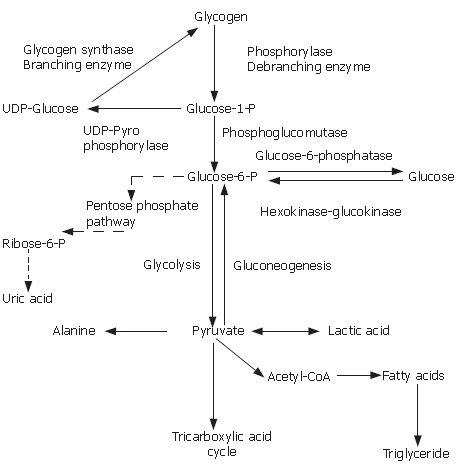
Glycogen storage diseases (GSD) are inherited metabolic disorders of glycogen metabolism. Different hormones, including insulin, glucagon, and cortisol regulate the relationship of glycolysis, gluconeogenesis and glycogen synthesis. The overall GSD incidence is estimated 1 case per 20000-43000 live births. There are over 12 types and they are classified based on the enzyme deficiency and the affected tissue. Disorders of glycogen degradation may affect primarily the liver, the muscle, or both. Type Ia involves the liver, kidney and intestine (and Ib also leukocytes), and the clinical manifestations are hepatomegaly, failure to thrive, hypoglycemia, hyperlactatemia, hyperuricemia and hyperlipidemia. Type IIIa involves both the liver and muscle, and IIIb solely the liver. The liver symptoms generally improve with age. Type IV usually presents in the first year of life, with hepatomegaly and growth retardation. The disease in general is progressive to cirrhosis. Type VI and IX are a heterogeneous group of diseases caused by a deficiency of the liver phosphorylase and phosphorylase kinase system. There is no hyperuricemia or hyperlactatemia. Type XI is characterized by hepatic glycogenosis and renal Fanconi syndrome. Type II is a prototype of inborn lysosomal storage diseases and involves many organs but primarily the muscle. Types V and VII involve only the muscle.
Figures
References
-
- Roach PJ. Glycogen and its metabolism. Curr Mol Med. 2002;2:101–120. - PubMed
-
- Saltik IN, Ozen H, Ciliv G, Koçak N, Yüce A, Gürakan F, Dinler G. Glycogen storage disease type Ia: frequency and clinical course in Turkish children. Indian J Pediatr. 2000;67:497–501. - PubMed
-
- Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics. 2000;105:e10. - PubMed
-
- Chen YT. Glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, Vale D, Childs B, et al., editors. The metabolic & molecular basis of inherited diseases. New York: McGraw-Hill; 2001. pp. 1521–1552.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
