

Amyloid beta ion channel: 3D structure and relevance to amyloid channel paradigm
- PMID: 17553456
- PMCID: PMC2692960
- DOI: 10.1016/j.bbamem.2007.04.021
Amyloid beta ion channel: 3D structure and relevance to amyloid channel paradigm
Abstract
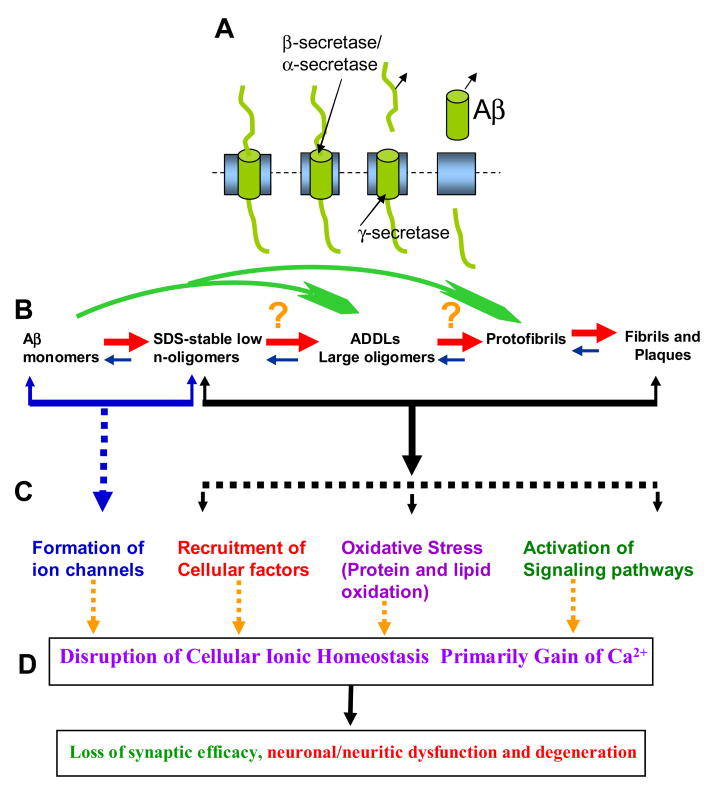
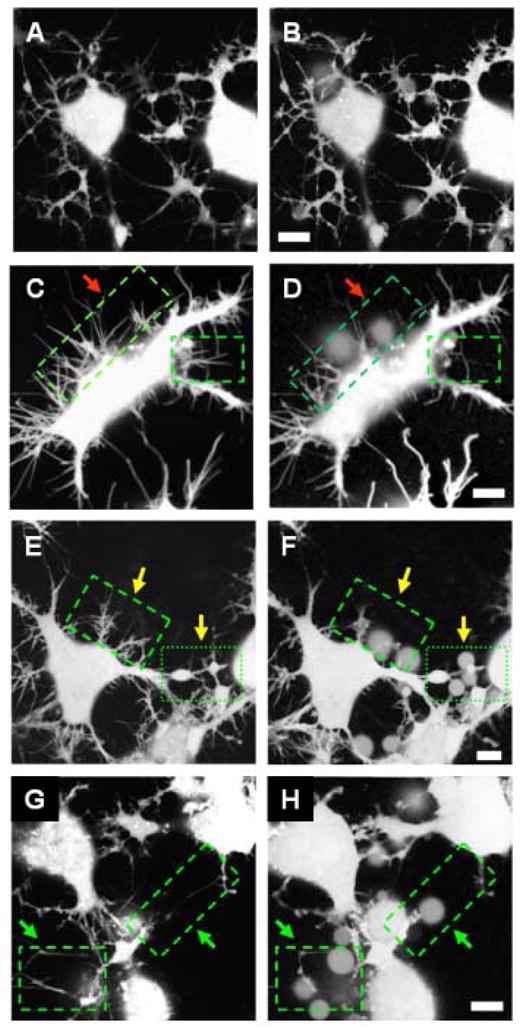
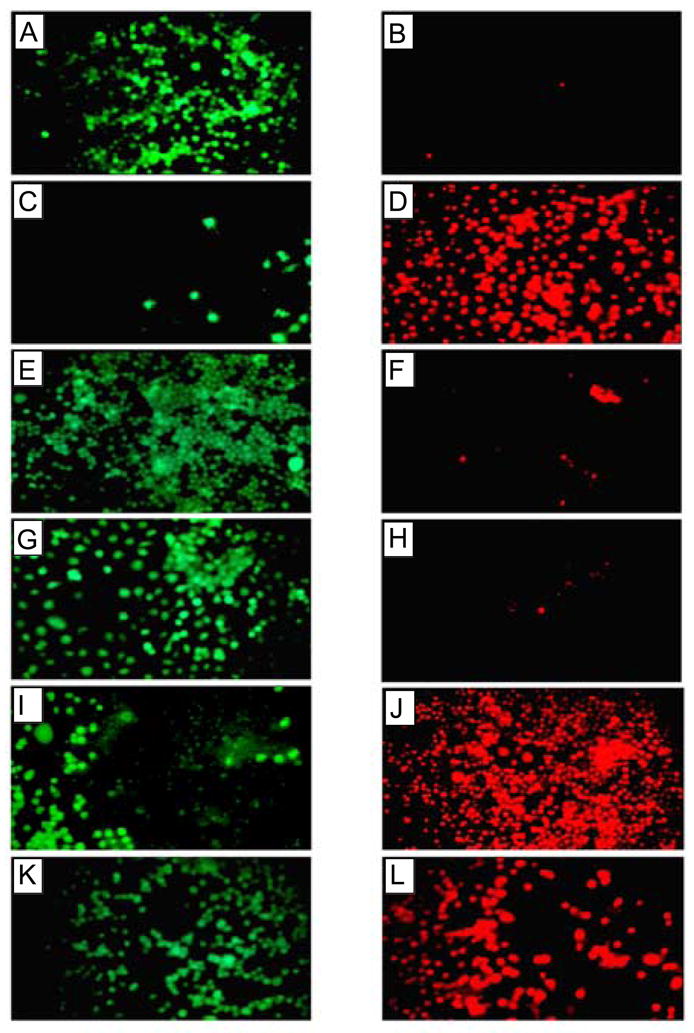
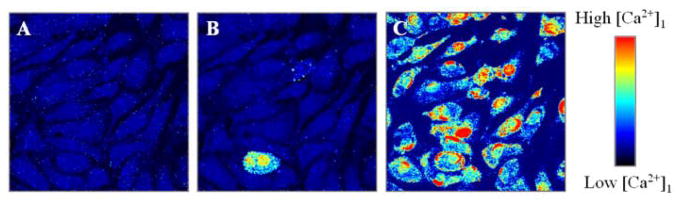
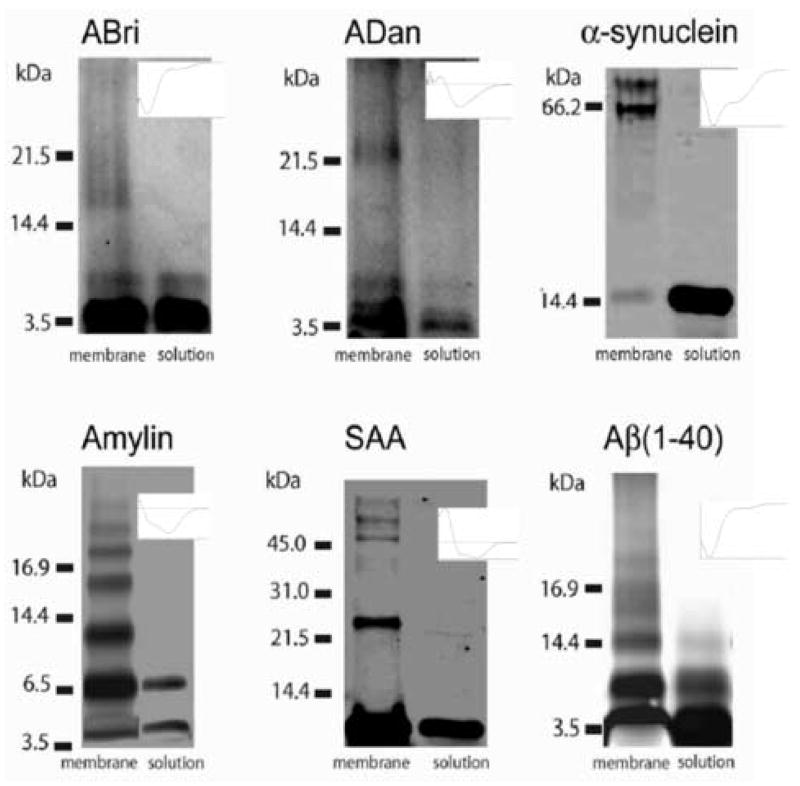
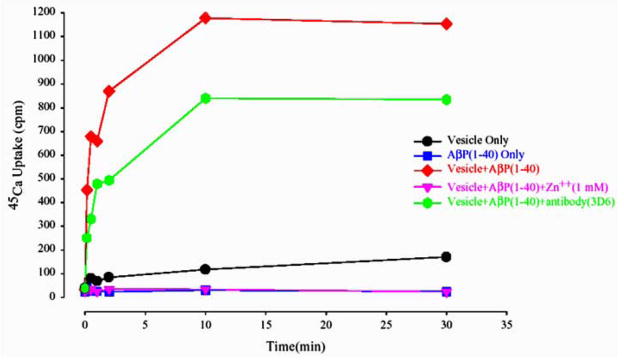
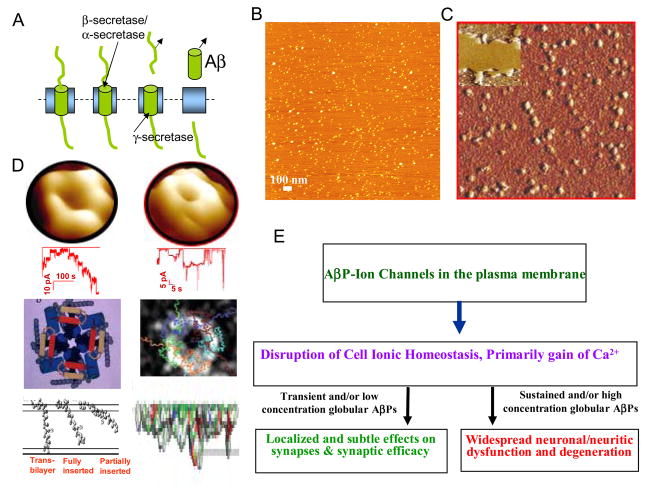
Alzheimer's disease (AD) is a protein misfolding disease. Early hypothesis of AD pathology posits that 39-43 AA long misfolded amyloid beta (Abeta) peptide forms a fibrillar structure and induces pathophysiological response by destabilizing cellular ionic homeostasis. Loss of cell ionic homeostasis is believed to be either indirectly due to amyloid beta-induced oxidative stress or directly by its interaction with the cell membrane and/or activating pathways for ion exchange. Significantly though, no Abeta specific cell membrane receptors are known and oxidative stress mediated pathology is only partial and indirect. Most importantly, recent studies strongly indicate that amyloid fibrils may not by themselves cause AD pathology. Subsequently, a competing hypothesis has been proposed wherein amyloid derived diffusible ligands (ADDLs) that are large Abeta oligomers (approximately >60 kDa), mediate AD pathology. No structural details, however, of these large globular units exist nor is there any known suitable mechanism by which they would induce AD pathology. Experimental data indicate that they alter cell viability by non-specifically changing the plasma membrane stability and increasing the overall ionic leakiness. The relevance of this non-specific mechanism for AD-specific pathology seems limited. Here, we provide a viable new paradigm: AD pathology mediated by amyloid ion channels made of small Abeta oligomers (trimers to octamers). This review is focused to 3D structural analysis of the Abeta channel. The presence of amyloid channels is consistent with electrophysiological and cell biology studies summarized in companion reviews in this special issue. They show ion channel-like activity and channel-mediated cell toxicity. Amyloid ion channels with defined gating and pharmacological agents would provide a tangible target for designing therapeutics for AD pathology.
Figures




Similar articles
-
Abeta ion channels. Prospects for treating Alzheimer's disease with Abeta channel blockers.Biochim Biophys Acta. 2007 Aug;1768(8):1952-65. doi: 10.1016/j.bbamem.2007.03.014. Epub 2007 Mar 24. Biochim Biophys Acta. 2007. PMID: 17490607 Review.
-
Atomic force microscopy and MD simulations reveal pore-like structures of all-D-enantiomer of Alzheimer's β-amyloid peptide: relevance to the ion channel mechanism of AD pathology.J Phys Chem B. 2012 Feb 9;116(5):1728-35. doi: 10.1021/jp2108126. Epub 2012 Jan 25. J Phys Chem B. 2012. PMID: 22217000 Free PMC article.
-
Amyloid β Ion Channels in a Membrane Comprising Brain Total Lipid Extracts.ACS Chem Neurosci. 2017 Jun 21;8(6):1348-1357. doi: 10.1021/acschemneuro.7b00006. Epub 2017 Feb 20. ACS Chem Neurosci. 2017. PMID: 28135799 Free PMC article.
-
Ion Channel Formation by Amyloid-β42 Oligomers but Not Amyloid-β40 in Cellular Membranes.J Biol Chem. 2017 Jan 27;292(4):1404-1413. doi: 10.1074/jbc.M116.762526. Epub 2016 Dec 7. J Biol Chem. 2017. PMID: 27927987 Free PMC article.
-
Disordered amyloidogenic peptides may insert into the membrane and assemble into common cyclic structural motifs.Chem Soc Rev. 2014 Oct 7;43(19):6750-64. doi: 10.1039/c3cs60459d. Chem Soc Rev. 2014. PMID: 24566672 Free PMC article. Review.
Cited by
-
Probing the Structure of Toxic Amyloid-β Oligomers with Electron Spin Resonance and Molecular Modeling.ACS Chem Neurosci. 2021 Apr 7;12(7):1150-1161. doi: 10.1021/acschemneuro.0c00714. Epub 2021 Mar 16. ACS Chem Neurosci. 2021. PMID: 33724783 Free PMC article.
-
Alzheimer Aβ peptide interactions with lipid membranes: fibrils, oligomers and polymorphic amyloid channels.Prion. 2012 Sep-Oct;6(4):339-45. doi: 10.4161/pri.21022. Epub 2012 Aug 9. Prion. 2012. PMID: 22874669 Free PMC article.
-
The on-fibrillation-pathway membrane content leakage and off-fibrillation-pathway lipid mixing induced by 40-residue β-amyloid peptides in biologically relevant model liposomes.Biochim Biophys Acta Biomembr. 2018 Sep;1860(9):1670-1680. doi: 10.1016/j.bbamem.2018.03.008. Epub 2018 Mar 13. Biochim Biophys Acta Biomembr. 2018. PMID: 29548698 Free PMC article.
-
Platelets are responsible for the accumulation of β-amyloid in blood clots inside and around blood vessels in mouse brain after thrombosis.Brain Res Bull. 2017 Jan;128:98-105. doi: 10.1016/j.brainresbull.2016.11.008. Epub 2016 Nov 28. Brain Res Bull. 2017. PMID: 27908798 Free PMC article.
-
Neurometals in the Pathogenesis of Prion Diseases.Int J Mol Sci. 2021 Jan 28;22(3):1267. doi: 10.3390/ijms22031267. Int J Mol Sci. 2021. PMID: 33525334 Free PMC article. Review.
References
-
- Dobson M. Protein folding and misfolding. Nature. 2003;426:884–890. - PubMed
-
- Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL. Cerebral amyloid angiopathies: A pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol. 2003;62:885–898. - PubMed
-
- Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. - PubMed
-
- Pallito MM, Ghanta J, Heinzelman P, Kiessling LL, Murphy RM. Recognition sequence design for peptidyl modulators of beta-amyloid aggregation and toxicity. Biochemistry. 1999;38:3570–3578. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
