

Mapping the human plasma proteome by SCX-LC-IMS-MS
- PMID: 17553692
- PMCID: PMC2195767
- DOI: 10.1016/j.jasms.2007.04.012
Mapping the human plasma proteome by SCX-LC-IMS-MS
Abstract
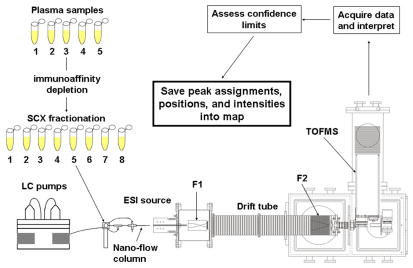
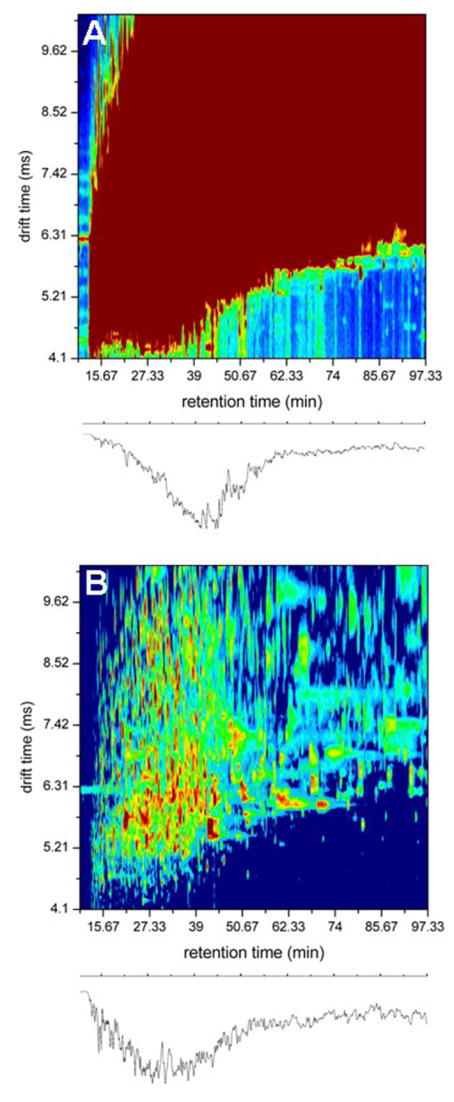
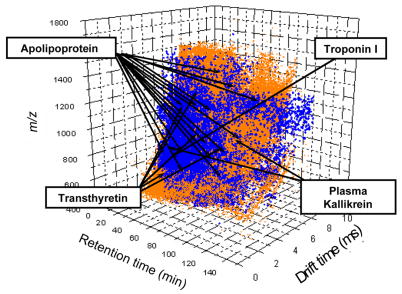
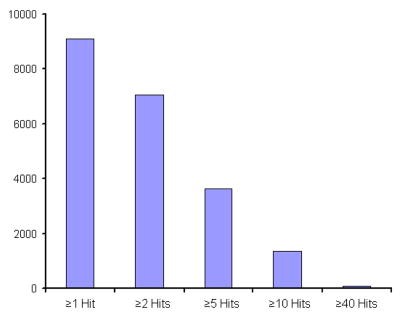
The advent of on-line multidimensional liquid chromatography-mass spectrometry has significantly impacted proteomic analyses of complex biological fluids such as plasma. However, there is general agreement that additional advances to enhance the peak capacity of such platforms are required to enhance the accuracy and coverage of proteome maps of such fluids. Here, we describe the combination of strong-cation-exchange and reversed-phase liquid chromatographies with ion mobility and mass spectrometry as a means of characterizing the complex mixture of proteins associated with the human plasma proteome. The increase in separation capacity associated with inclusion of the ion mobility separation leads to generation of one of the most extensive proteome maps to date. The map is generated by analyzing plasma samples of five healthy humans; we report a preliminary identification of 9087 proteins from 37,842 unique peptide assignments. An analysis of expected false-positive rates leads to a high-confidence identification of 2928 proteins. The results are catalogued in a fashion that includes positions and intensities of assigned features observed in the datasets as well as pertinent identification information such as protein accession number, mass, and homology score/confidence indicators. Comparisons of the assigned features reported here with other datasets shows substantial agreement with respect to the first several hundred entries; there is far less agreement associated with detection of lower abundance components.
Figures


References
-
- Wasinger VC, Cordwell SJ, Cerpa-Poljak A, Yan JX, Gooley AA, Wilkins MR, Duncan MW, Harris R, Williams KL, Humphery-Smith I. Progress with Gene-Product Mapping of the Mollicutes: Mycoplasma Genitalium. Electrophoresis. 1995;16:1090–1094. - PubMed
-
-
For proteomic platform technology development information see the following reviews andreferences therein: Hancock WS, Apffel AJ, Chakel JA, Hahnenberger KC, Choudhary G, Traina J, Pungor E. Integrated Genomic/Proteomic Analysis. Anal Chem. 1999;71:742A–748A.Schweitzer B, Kingsmore SF. Measuring Proteins on Microarrays. Curr Opin Biotech. 2002;13:14–19.Figeys D. Proteomics in 2002: a Year of Technical Development and Wide-Ranging Applications. Anal Chem. 2003;75:2891–2905.Romijn EP, Krijgsveld J, Heck AJR. Recent Liquid Chromatographic-(Tandem) Mass Spectrometric Applications in Proteomics. J Chromat A. 2003;1000:589–608.Aebersold R, Mann M. Mass Spectrometry-Based Proteomics. Nature. 2003;422:198–207.Page JS, Masselon CD, Smith RD. FTICR Mass Spectrometry for Qualitative and Quantitative Bioanalyses. Curr Opin Biotech. 2004;15:3–11.Anderson L. Candidate-Based Proteomics in the Search for Biomarkers of Cardiovascular Disease. J Physiol-London. 2005;563:23–60.
-
-
- Anderson NL, Anderson NG. The Human Plasma Proteome: History, Character, and Diagnostic Prospects. Mol Cel Proteomics. 2002;1:845–867. - PubMed
-
- Liu X, Plasencia M, Ragg S, Valentine SJ, Clemmer DE. Development of High-Throughput Dispersive LC-Ion Mobility-TOFMS Techniques for Analyzing the Human Plasma Proteome. Brief Funct Genomics Proteomics. 2004;3:177–186. - PubMed
-
-
The latest version of the Swiss-Prot protein database can be found at the following web address: http://www.expasy.uniprot.org/database/knowledgebase.shtml.
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
