

Regulation of the NMDA receptor-mediated synaptic response by acetylcholinesterase inhibitors and its impairment in an animal model of Alzheimer's disease
- PMID: 17555845
- PMCID: PMC2613405
- DOI: 10.1016/j.neurobiolaging.2007.04.023
Regulation of the NMDA receptor-mediated synaptic response by acetylcholinesterase inhibitors and its impairment in an animal model of Alzheimer's disease
Abstract
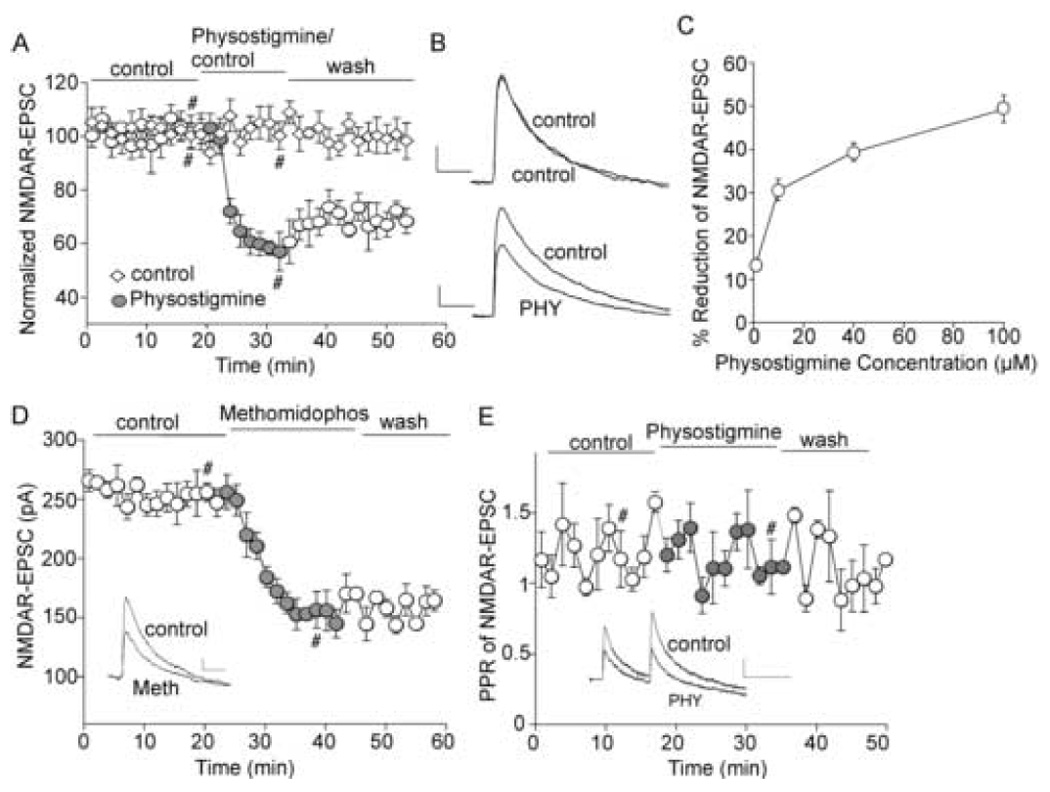
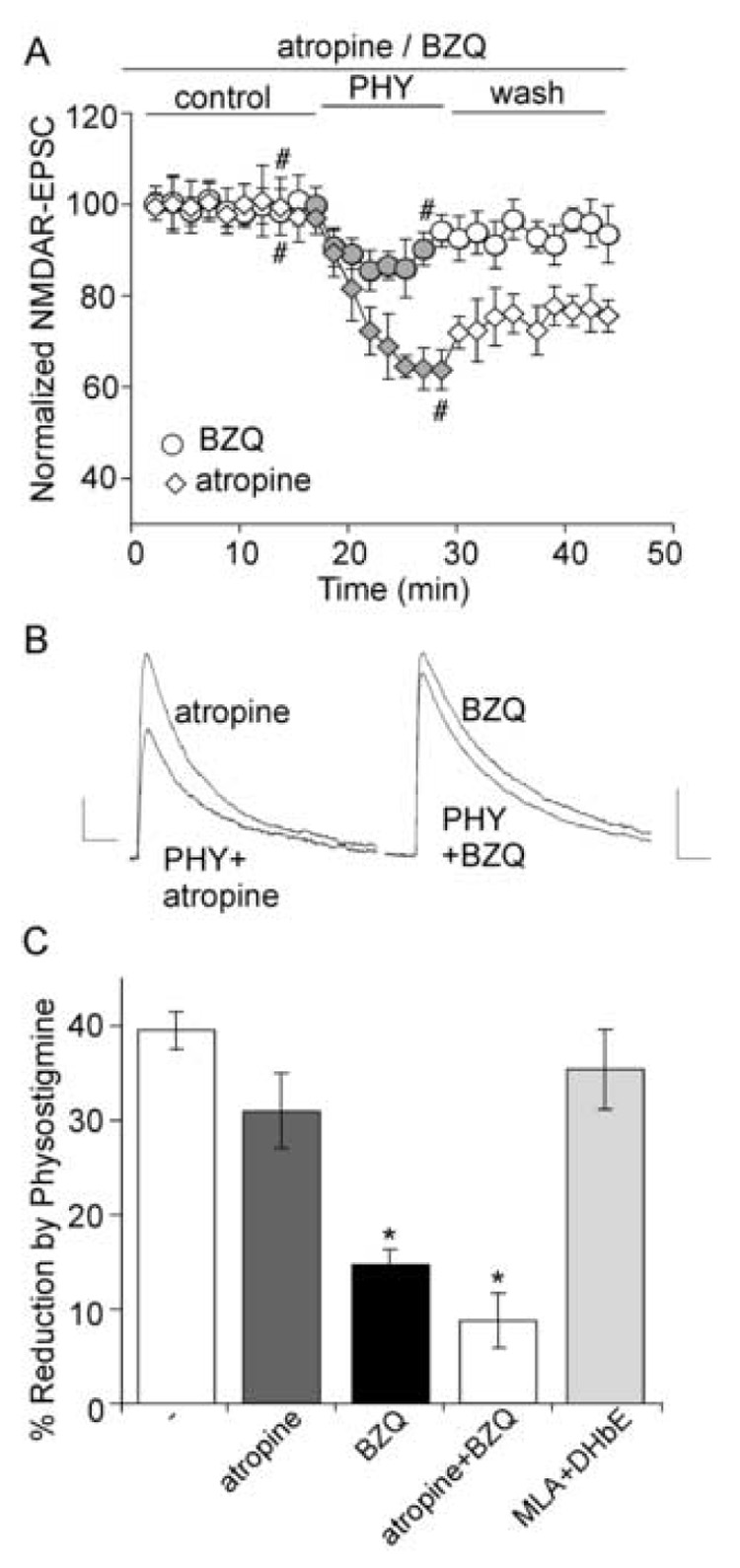
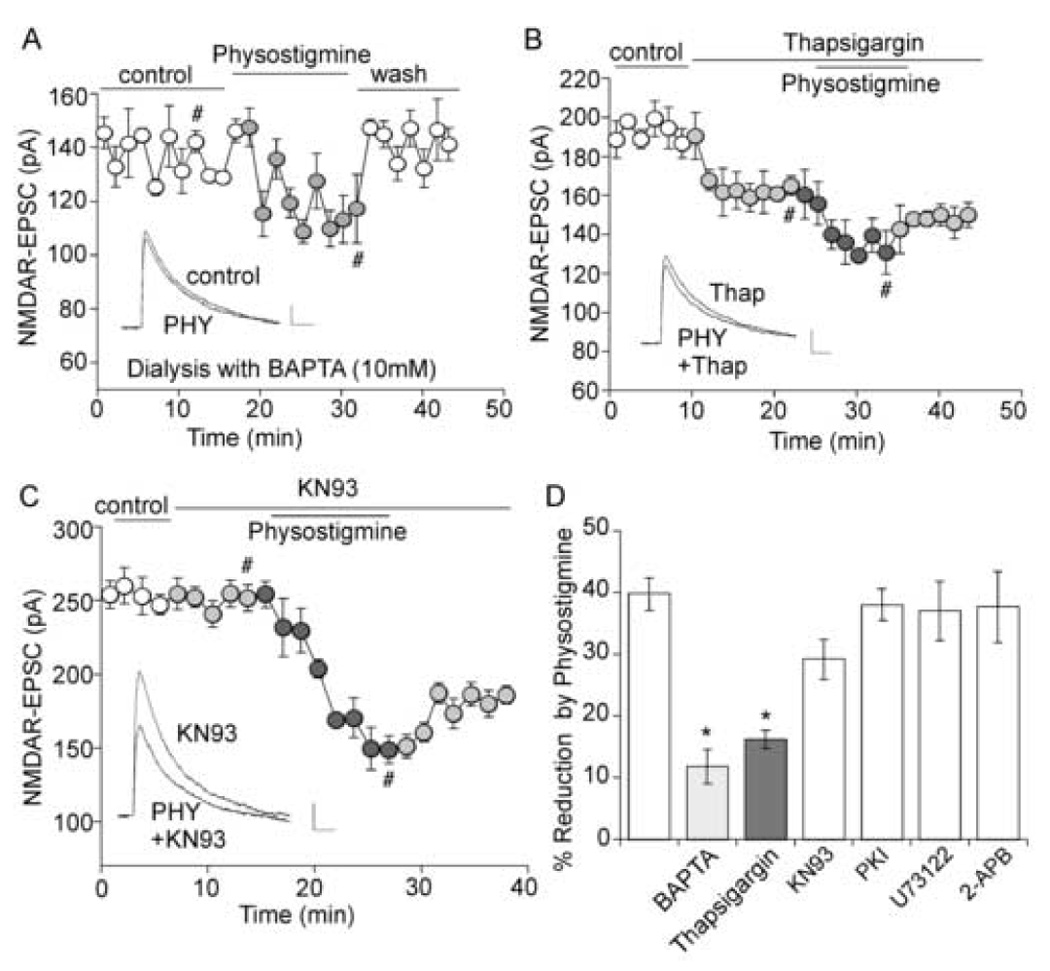
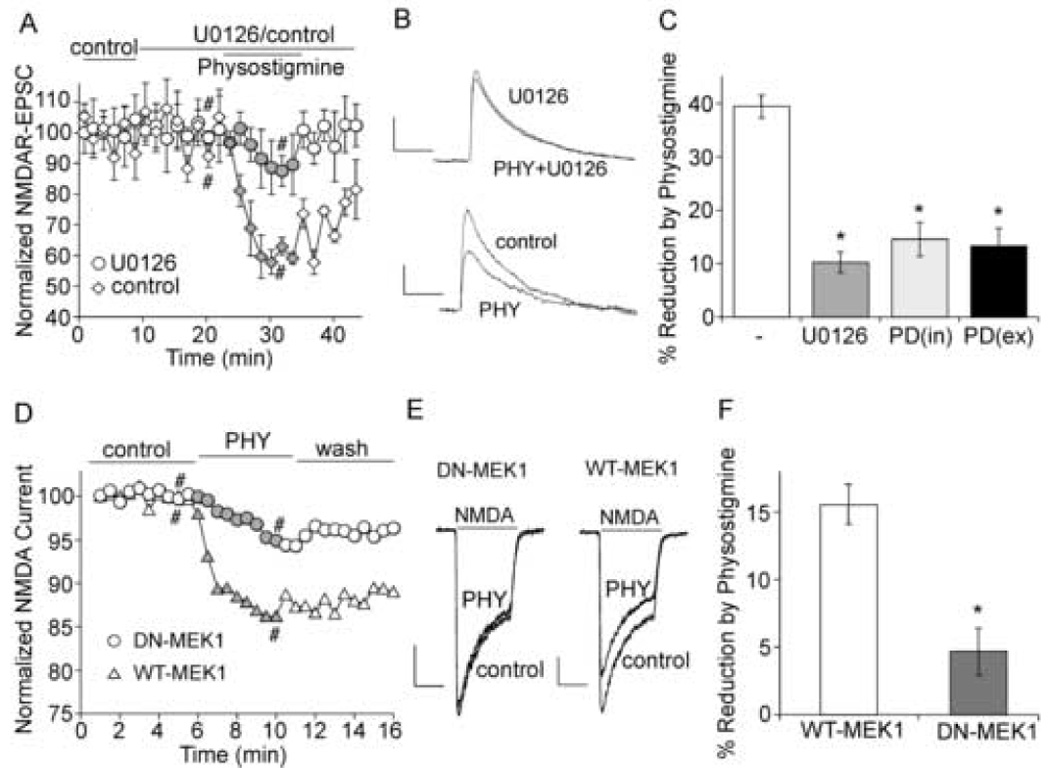
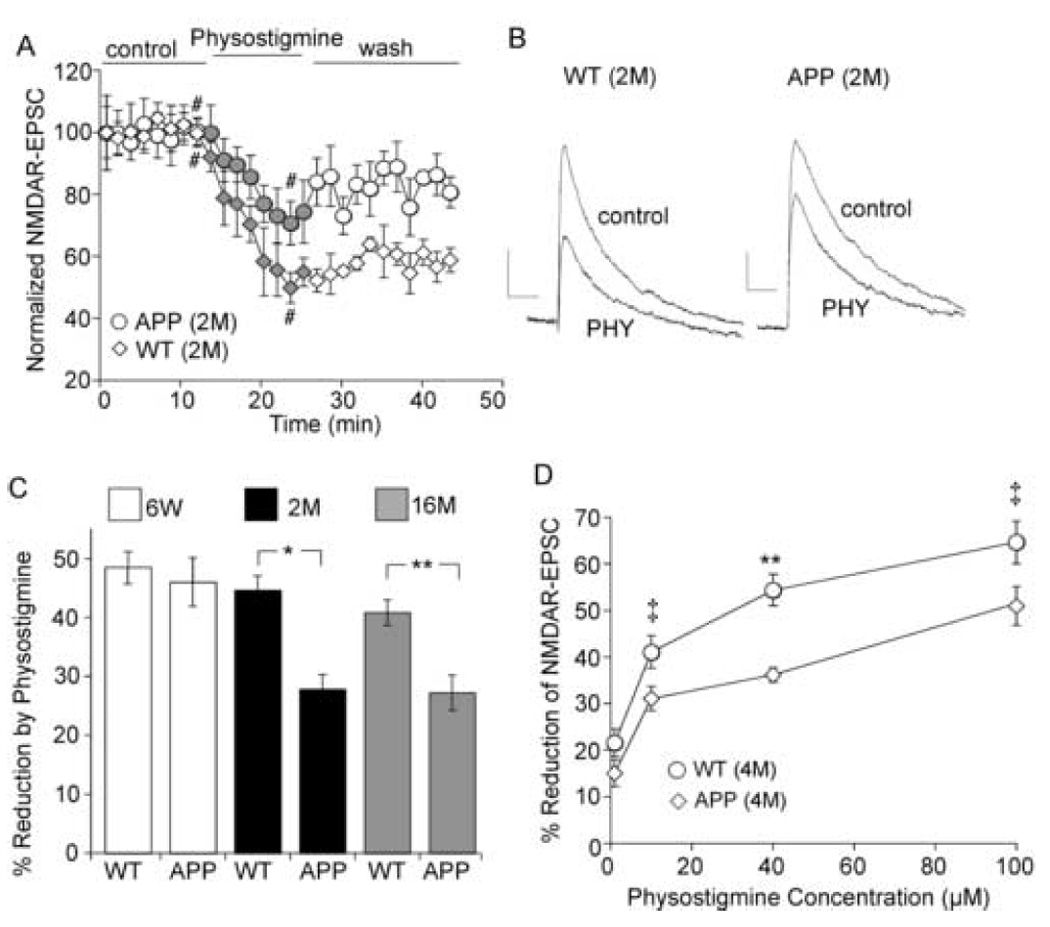
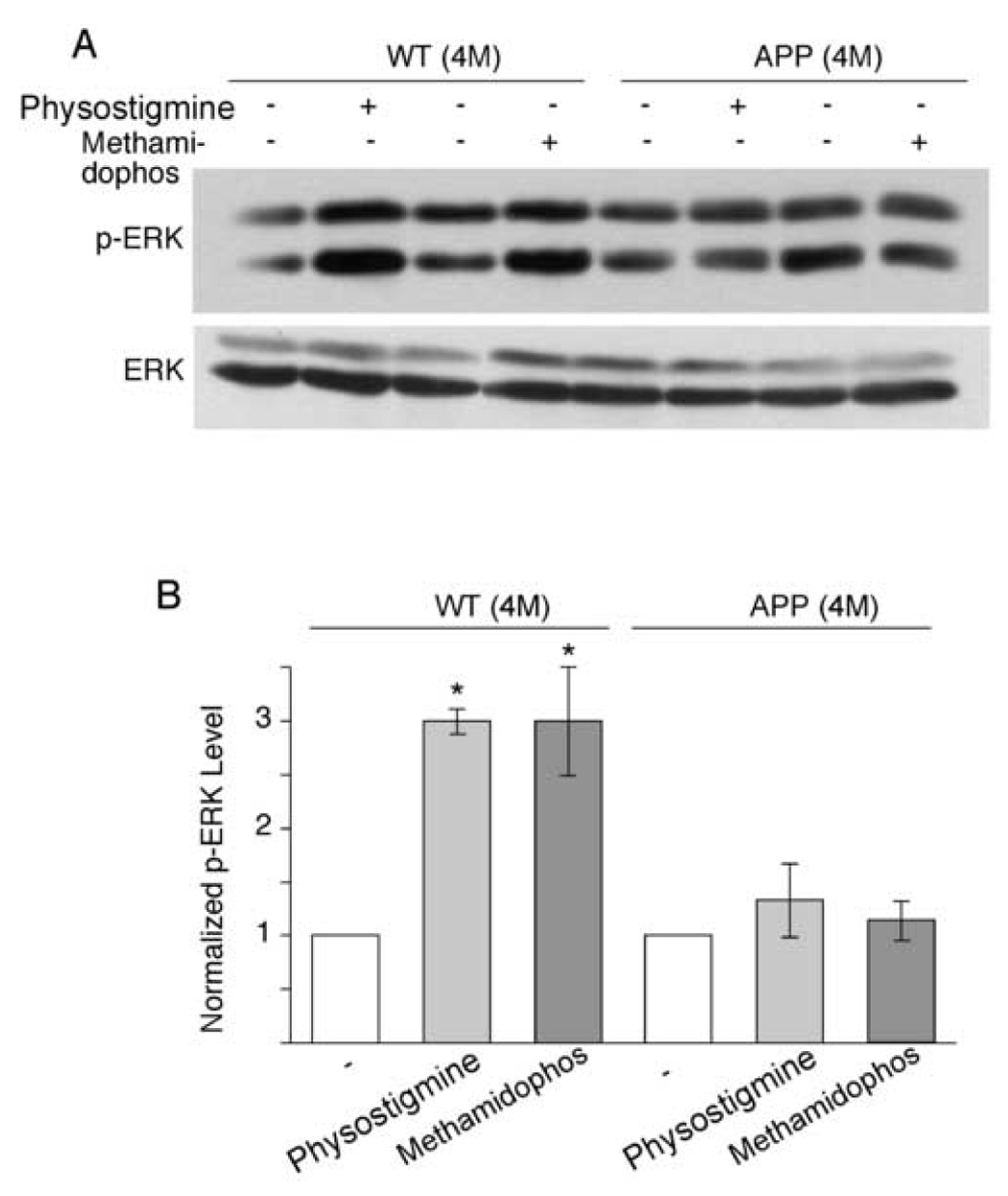
The cholinergic system is crucial for cognitive processes and the deficient acetylcholine (ACh) function has been implicated in Alzheimer's disease (AD). Inhibitors of acetylcholinesterase (AChE), which act to enhance cholinergic function by prolonging the action of endogenously released ACh, have been used as the major therapy of AD. To understand the functional roles of cholinergic enhancement in prefrontal cortex (PFC), a key brain region for cognition, we examined the impact of AChE inhibitors in PFC neurons on synaptic responses mediated by the NMDA receptor (NMDAR), an important player in learning and memory. We found that AChE inhibitors produced a strong and persistent reduction of the amplitude of NMDA receptor-mediated excitatory postsynaptic current (NMDAR-EPSC). This effect was mainly mediated by nicotinic ACh receptors, and through a Ca(2+)-dependent mechanism. Inhibition of extracellular signal-regulated kinases (ERK) abolished the regulation of NMDAR function by AChE inhibitors, suggesting the involvement of ERK. In the transgenic mouse model of AD overexpressing mutant beta-amyloid precursor protein (APP), the effect of AChE inhibitors on NMDAR-EPSC was significantly impaired, which was associated with their diminished effect on ERK activation. Taken together, these results suggest that one of the key targets of endogenous ACh involved in cognition is the NMDAR-mediated transmission. Loss of the regulation of synaptic NMDAR responses by endogenous ACh may contribute to the cognitive deficiency in AD.
Conflict of interest statement
All authors have no actual or potential conflicts of interest with other people or organizations within three years of beginning the work presented here.
Figures
References
-
- Akasofu S, Kimura M, Kosasa T, Ogura H, Sawada K. Protective effect of donepezil in primary-cultured rat cortical neurons exposed to N-methyl-d-aspartate (NMDA) toxicity. Eur J Pharmacol. 2006;530(3):215–222. - PubMed
-
- Auld DS, Kar S, Quirion R. Beta-amyloid peptides as direct cholinergic neuromodulators: a missing link? Trends Neurosci. 1998;21(1):43–49. - PubMed
-
- Bartus RT, Dean RL, 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217(4558):408–414. - PubMed
-
- Benzi G, Moretti A. Is there a rationale for the use of acetylcholinesterase inhibitors in the therapy of Alzheimer's disease? Eur J Pharmacol. 1998;346(1):1–13. - PubMed
-
- Blusztajn JK, Berse B. The cholinergic neuronal phenotype in Alzheimer's disease. Metab Brain Dis. 2000;15(1):45–64. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
