

Production of recombinant beta-hexosaminidase A, a potential enzyme for replacement therapy for Tay-Sachs and Sandhoff diseases, in the methylotrophic yeast Ogataea minuta
- PMID: 17557860
- PMCID: PMC1951009
- DOI: 10.1128/AEM.00463-07
Production of recombinant beta-hexosaminidase A, a potential enzyme for replacement therapy for Tay-Sachs and Sandhoff diseases, in the methylotrophic yeast Ogataea minuta
Abstract
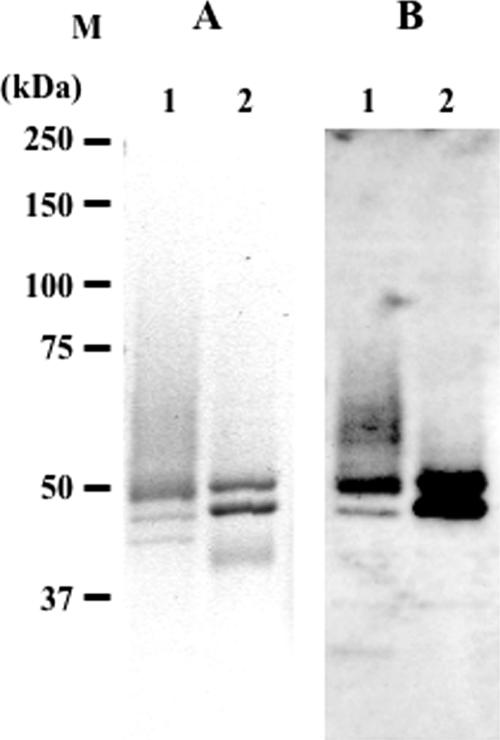
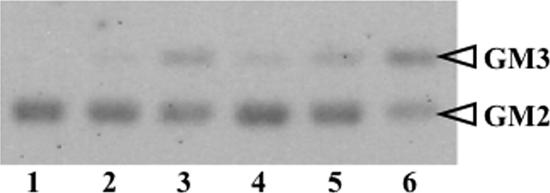
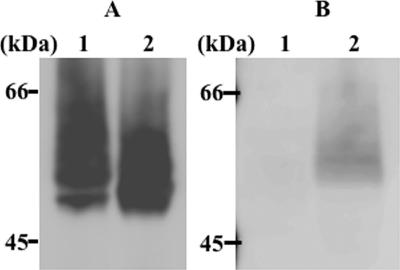
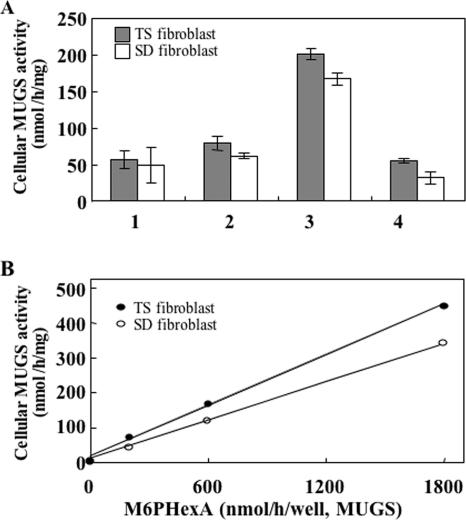
Human beta-hexosaminidase A (HexA) is a heterodimeric glycoprotein composed of alpha- and beta-subunits that degrades GM2 gangliosides in lysosomes. GM2 gangliosidosis is a lysosomal storage disease in which an inherited deficiency of HexA causes the accumulation of GM2 gangliosides. In order to prepare a large amount of HexA for a treatment based on enzyme replacement therapy (ERT), recombinant HexA was produced in the methylotrophic yeast Ogataea minuta instead of in mammalian cells, which are commonly used to produce recombinant enzymes for ERT. The problem of antigenicity due to differences in N-glycan structures between mammalian and yeast glycoproteins was potentially resolved by using alpha-1,6-mannosyltransferase-deficient (och1Delta) yeast as the host. Genes encoding the alpha- and beta-subunits of HexA were integrated into the yeast cell, and the heterodimer was expressed together with its isozymes HexS (alphaalpha) and HexB (betabeta). A total of 57 mg of beta-hexosaminidase isozymes, of which 13 mg was HexA (alphabeta), was produced per liter of medium. HexA was purified with immobilized metal affinity column for the His tag attached to the beta-subunit. The purified HexA was treated with alpha-mannosidase to expose mannose-6-phosphate (M6P) residues on the N-glycans. The specific activities of HexA and M6P-exposed HexA (M6PHexA) for the artificial substrate 4MU-GlcNAc were 1.2 +/- 0.1 and 1.7 +/- 0.3 mmol/h/mg, respectively. The sodium dodecyl sulfate-polyacrylamide gel electrophoresis pattern suggested a C-terminal truncation in the beta-subunit of the recombinant protein. M6PHexA was incorporated dose dependently into GM2 gangliosidosis patient-derived fibroblasts via M6P receptors on the cell surface, and degradation of accumulated GM2 ganglioside was observed.
Figures

Similar articles
-
Inefficiency in GM2 ganglioside elimination by human lysosomal beta-hexosaminidase beta-subunit gene transfer to fibroblastic cell line derived from Sandhoff disease model mice.Biol Pharm Bull. 2006 Aug;29(8):1564-9. doi: 10.1248/bpb.29.1564. Biol Pharm Bull. 2006. PMID: 16880605
-
[Molecular pathogenesis and therapeutic approach of GM2 gangliosidosis].Yakugaku Zasshi. 2013;133(2):269-74. doi: 10.1248/yakushi.12-00199. Yakugaku Zasshi. 2013. PMID: 23370522 Review. Japanese.
-
Promoters for the human beta-hexosaminidase genes, HEXA and HEXB.DNA Cell Biol. 1996 Feb;15(2):89-97. doi: 10.1089/dna.1996.15.89. DNA Cell Biol. 1996. PMID: 8634145
-
Introduction of an N-glycan sequon into HEXA enhances human beta-hexosaminidase cellular uptake in a model of Sandhoff disease.Mol Ther. 2010 Aug;18(8):1519-26. doi: 10.1038/mt.2010.113. Epub 2010 Jun 22. Mol Ther. 2010. PMID: 20571546 Free PMC article.
-
The biochemistry of HEXA and HEXB gene mutations causing GM2 gangliosidosis.Biochim Biophys Acta. 1991 Feb 22;1096(2):87-94. doi: 10.1016/0925-4439(91)90044-a. Biochim Biophys Acta. 1991. PMID: 1825792 Review. No abstract available.
Cited by
-
Yeast-Produced Human Recombinant Lysosomal β-Hexosaminidase Efficiently Rescues GM2 Ganglioside Accumulation in Tay-Sachs Disease.J Pers Med. 2025 May 10;15(5):196. doi: 10.3390/jpm15050196. J Pers Med. 2025. PMID: 40423067 Free PMC article.
-
The MFα signal sequence in yeast-based protein secretion: challenges and innovations'.Appl Microbiol Biotechnol. 2025 Jun 5;109(1):138. doi: 10.1007/s00253-025-13532-z. Appl Microbiol Biotechnol. 2025. PMID: 40471355 Free PMC article. Review.
-
New Approaches to Tay-Sachs Disease Therapy.Front Physiol. 2018 Nov 20;9:1663. doi: 10.3389/fphys.2018.01663. eCollection 2018. Front Physiol. 2018. PMID: 30524313 Free PMC article. Review.
-
Lysosomal enzyme replacement therapies: Historical development, clinical outcomes, and future perspectives.Adv Drug Deliv Rev. 2017 Sep 1;118:109-134. doi: 10.1016/j.addr.2017.05.004. Epub 2017 May 11. Adv Drug Deliv Rev. 2017. PMID: 28502768 Free PMC article. Review.
-
Unusual case of Juvenile Tay-Sachs disease.BMJ Case Rep. 2019 Sep 12;12(9):e230140. doi: 10.1136/bcr-2019-230140. BMJ Case Rep. 2019. PMID: 31519716 Free PMC article.
References
-
- Amalfitano, A., A. J. McVie-Wylie, H. Hu, T. L. Dawson, N. Rabens, P. Plotz, and Y. T. Chen. 1999. Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid-α-glucosidase. Proc. Natl. Acad. Sci. USA 96:8861-8866. - PMC - PubMed
-
- Andersson, U., D. Smith, M. Jeyakumar, T. D. Butters, M. C. Borja, R. A. Dwek, and F. M. Platt. 2004. Improved outcome of N-butyldeoxygalactonojirimycin-mediated substrate reduction therapy in a mouse model of Sandhoff disease. Neurobiol. Dis. 16:506-515. - PubMed
-
- Asano, N., S. Ishii, H. Kizu, K. Ikeda, K. Yasuda, A. Kato, O. R. Maritn, and J. Q. Fan. 2000. In vivo inhibition and intracellular enhancement of lysosomal α-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur. J. Biochem. 267:4179-4186. - PubMed
-
- Barton, N. W., R. O. Brady, J. M. Dambrosia, A. M. Di Bisceglie, S. H. Doppelt, S. C. Hill, H. J. Mankin, G. J. Murray, R. I. Parker, and C. E. Argoff. 1991. Replacement therapy for inherited enzyme deficiency-macrophage-medicated glucocerebrosidase for Gaucher's disease. N. Engl. J. Med. 324:1464-1470. - PubMed
-
- Butters, T. D., R. A. Dwek, and F. M. Platt. 2000. Inhibition of glycosphingolipid biosynthesis: application to lysosomal storage disorders. Chem. Rev. 100:4683-4696. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
