

Improved benchmarks for computational motif discovery
- PMID: 17559676
- PMCID: PMC1903367
- DOI: 10.1186/1471-2105-8-193
Improved benchmarks for computational motif discovery
Abstract
Background: An important step in annotation of sequenced genomes is the identification of transcription factor binding sites. More than a hundred different computational methods have been proposed, and it is difficult to make an informed choice. Therefore, robust assessment of motif discovery methods becomes important, both for validation of existing tools and for identification of promising directions for future research.
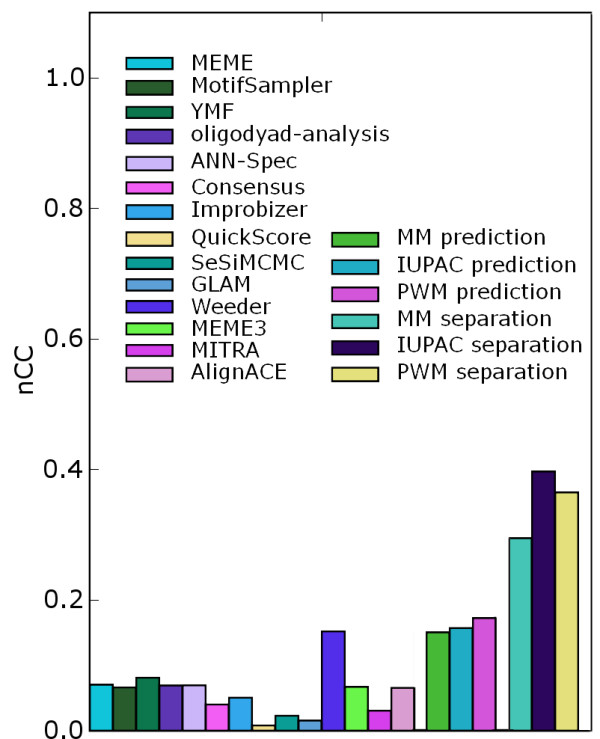
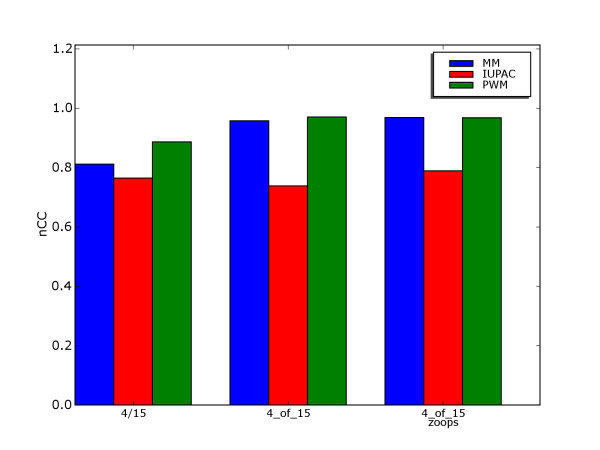
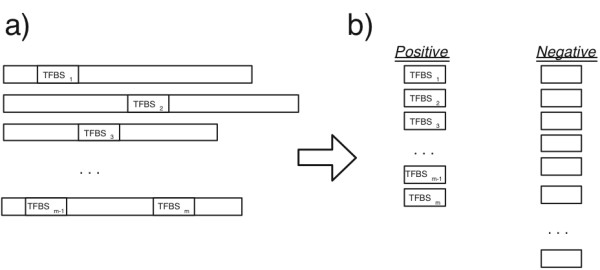
Results: We use a machine learning perspective to analyze collections of transcription factors with known binding sites. Algorithms are presented for finding position weight matrices (PWMs), IUPAC-type motifs and mismatch motifs with optimal discrimination of binding sites from remaining sequence. We show that for many data sets in a recently proposed benchmark suite for motif discovery, none of the common motif models can accurately discriminate the binding sites from remaining sequence. This may obscure the distinction between the potential performance of the motif discovery tool itself versus the intrinsic complexity of the problem we are trying to solve. Synthetic data sets may avoid this problem, but we show on some previously proposed benchmarks that there may be a strong bias towards a presupposed motif model. We also propose a new approach to benchmark data set construction. This approach is based on collections of binding site fragments that are ranked according to the optimal level of discrimination achieved with our algorithms. This allows us to select subsets with specific properties. We present one benchmark suite with data sets that allow good discrimination between positive and negative instances with the common motif models. These data sets are suitable for evaluating algorithms for motif discovery that rely on these models. We present another benchmark suite where PWM, IUPAC and mismatch motif models are not able to discriminate reliably between positive and negative instances. This suite could be used for evaluating more powerful motif models.
Conclusion: Our improved benchmark suites have been designed to differentiate between the performance of motif discovery algorithms and the power of motif models. We provide a web server where users can download our benchmark suites, submit predictions and visualize scores on the benchmarks.
Figures









References
-
- Hughes JD, Estep PW, Tavazoie S, Church GM. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol. 2000;296:1205–14. - PubMed
-
- Bailey TL, Elkan C. The value of prior knowledge in discovering motifs with MEME. Proc Int Conf Intell Syst Mol Biol. 1995;3:21–9. - PubMed
-
- Marsan L, Sagot MF. RECOMB '00: Proceedings of the fourth annual international conference on Computational molecular biology. New York, NY, USA: ACM Press; 2000. Extracting structured motifs using a suffix tree-algorithms and application to promoter consensus identification; pp. 210–219. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
