

Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H
- PMID: 17564959
- PMCID: PMC1950914
- DOI: 10.1086/518428
Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot-Marie-Tooth type 4H
Abstract
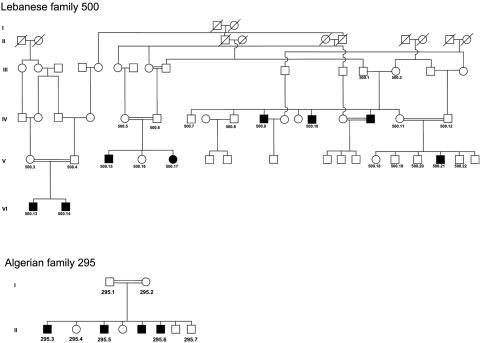
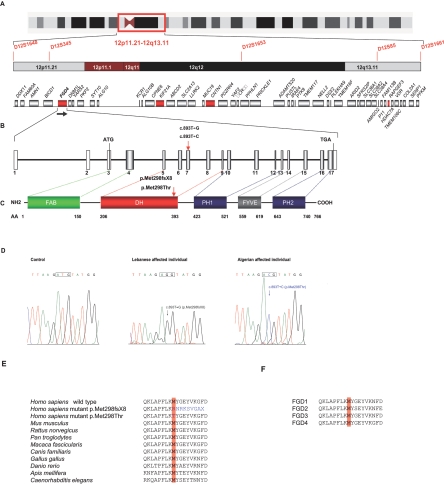
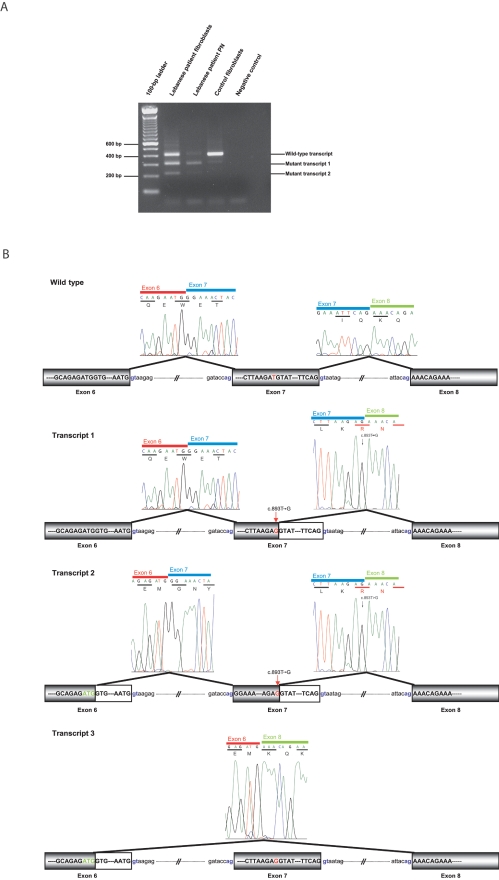
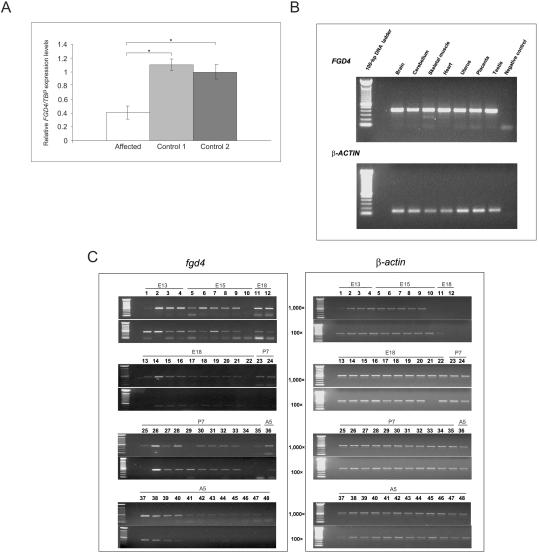
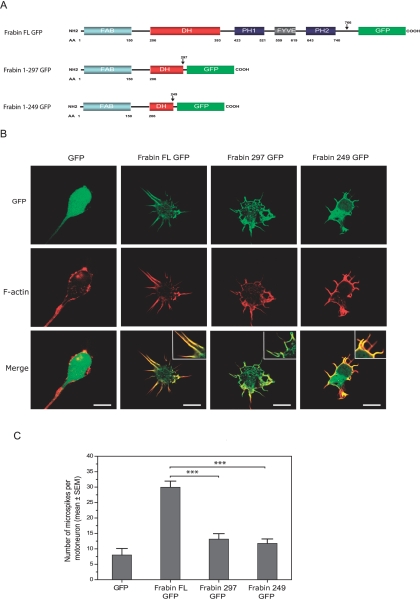
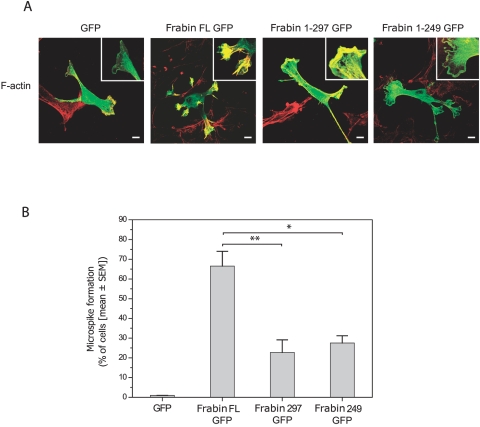
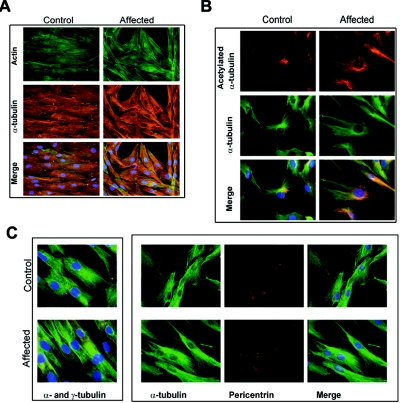
Charcot-Marie-Tooth (CMT) disorders are a clinically and genetically heterogeneous group of hereditary motor and sensory neuropathies characterized by muscle weakness and wasting, foot and hand deformities, and electrophysiological changes. The CMT4H subtype is an autosomal recessive demyelinating form of CMT that was recently mapped to a 15.8-Mb region at chromosome 12p11.21-q13.11, in two consanguineous families of Mediterranean origin, by homozygosity mapping. We report here the identification of mutations in FGD4, encoding FGD4 or FRABIN (FGD1-related F-actin binding protein), in both families. FRABIN is a GDP/GTP nucleotide exchange factor (GEF), specific to Cdc42, a member of the Rho family of small guanosine triphosphate (GTP)-binding proteins (Rho GTPases). Rho GTPases play a key role in regulating signal-transduction pathways in eukaryotes. In particular, they have a pivotal role in mediating actin cytoskeleton changes during cell migration, morphogenesis, polarization, and division. Consistent with these reported functions, expression of truncated FRABIN mutants in rat primary motoneurons and rat Schwann cells induced significantly fewer microspikes than expression of wild-type FRABIN. To our knowledge, this is the first report of mutations in a Rho GEF protein being involved in CMT.
Figures
References
Web Resources
-
- GenBank, http://www.ncbi.nlm.nih/Genbank/ (for KIF21A [accession number AY368076]; RAPGEF3 [accession number U78168]; FGD4 [accession number NM_139241]; FGD4 mRNA [accession numbers AK057294, BC045552, and AY367054] and ESTs for H. sapiens [accession number AA305646] and Macaca mulatta [accession number CN643653]; Fgd4 [accession number NM_139234]; FGD4 homologs FGD1 [accession number NP_004454], FGD2 [accession number NP_775829], and FGD3 [NP_149077]; and FRABIN proteins for H. sapiens [accession number NP_640334], Rattus norvegicus [accession number NP_640356], Mus musculus [accession number NP_631978], Macaca fascicularis [accession number BAE90450], Caenorhabditis elegans [accession number NP_001023039], Canis familiaris [accession number XP_543741], Pan troglodytes [accession number XP_520721], Gallus gallus [accession number XP_416365], Apis mellifera [accession number XP_394280], and Danio rerio [accession number CAK11116])
-
- Inherited Peripheral Neuropathies Mutation Database, http://www.molgen.ua.ac.be/CMTMutations/default.cfm
-
- Neuromuscular Disease Center, http://www.neuro.wustl.edu/neuromuscular/time/hmsn.html (for comprehensive and updated data about CMT diseases)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for GDAP1, CMT4A, MTMR2, CMT4B1, SBF2/MTMR13, CMT4B2, SH3CT2/KIAA1985, CMT4C, NDRG1, HMSN-Lom, EGR2, P0, PMP22, CMT4E, Dejerine-Sottas syndrome, PRX, CMT4F, HMSN-Russe, and CMT4H)
-
- ORF Finder, http://www.ncbi.nlm.nih.gov/gorf/gorf.html
References
-
- Skre H (1974) Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet 6:98–118 - PubMed
-
- Dyck PJ, Chance P, Lebo R, Carney AJ (1993) Hereditary motor and sensory neuropathies. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Podulso JF (eds) Peripheral neuropathy. WB Saunders, Philadelphia, pp 1094–1136
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
