

Identification of risk-related haplotypes with the use of multiple SNPs from nuclear families
- PMID: 17564963
- PMCID: PMC1950926
- DOI: 10.1086/518670
Identification of risk-related haplotypes with the use of multiple SNPs from nuclear families
Abstract
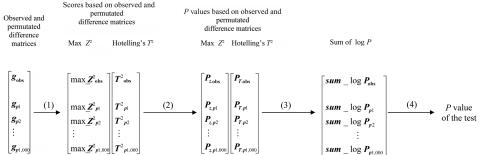
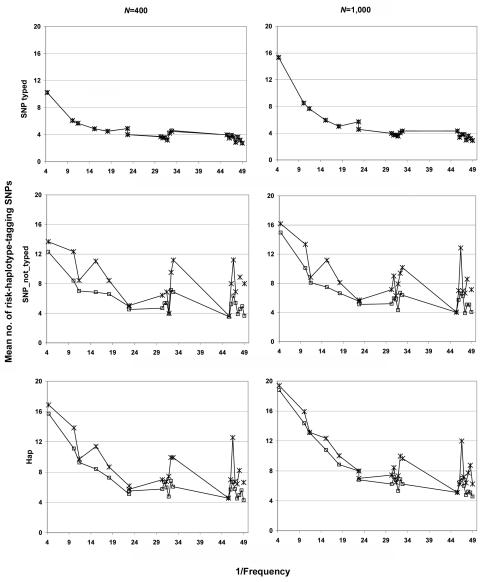
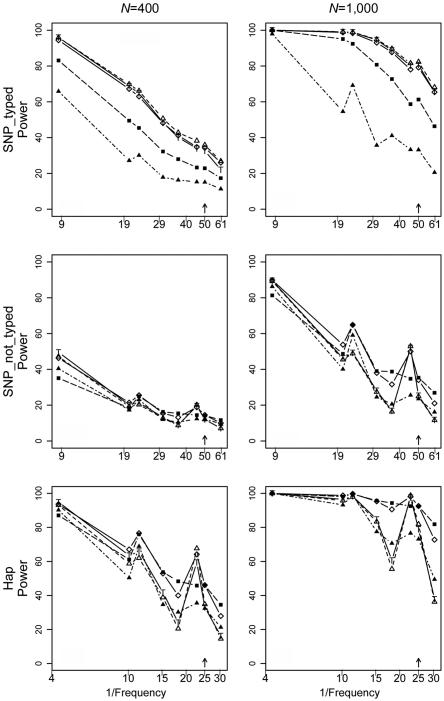
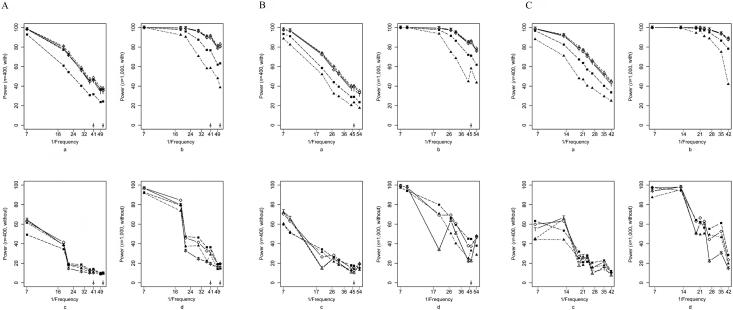
Family-based association studies offer robustness to population stratification and can provide insight into maternally mediated and parent-of-origin effects. Usually, such studies investigate multiple markers covering a gene or chromosomal region of interest. We propose a simple and general method to test the association of a disease trait with multiple, possibly linked SNP markers and, subsequently, to nominate a set of "risk-haplotype-tagging alleles." Our test, the max_Zeta(2) test, uses only the genotypes of affected individuals and their parents without requiring the user to either know or assign haplotypes and their phases. It also accommodates sporadically missing SNP data. In the spirit of the pedigree disequilibrium test, our procedure requires only a vector of differences with expected value 0 under the null hypothesis. To enhance power against a range of alternatives when genotype data are complete, we also consider a method for combining multiple tests; here, we combine max_Zeta(2) and Hotelling's Gamma(2). To facilitate discovery of risk-related haplotypes, we develop a simple procedure for nominating risk-haplotype-tagging alleles. Our procedures can also be used to study maternally mediated genetic effects and to explore imprinting. We compare the statistical power of several competing testing procedures through simulation studies of case-parents triads, whose diplotypes are simulated on the basis of draws from the HapMap-based known haplotypes of four genes. In our simulations, the max_Zeta(2) test and the max_TDT (transmission/disequilibrium test) proposed by McIntyre et al. perform almost identically, but max_Zeta(2), unlike max_TDT, extends directly to the investigation of maternal effects. As an illustration, we reanalyze data from a previously reported orofacial cleft study, to now investigate both fetal and maternal effects of the IRF6 gene.
Figures
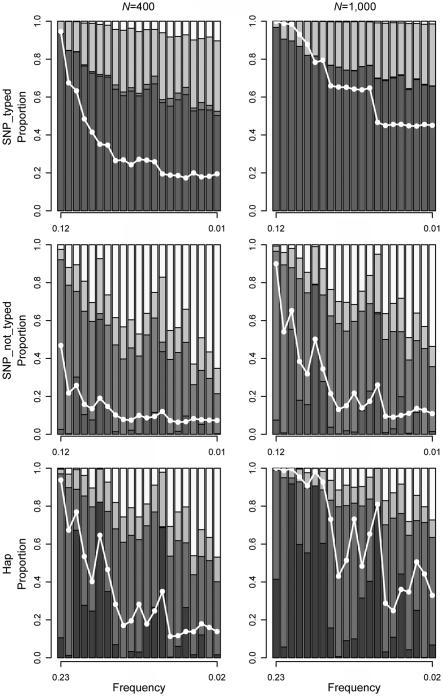
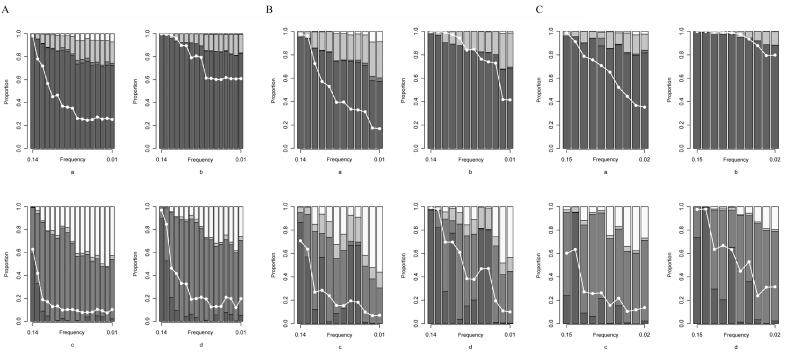
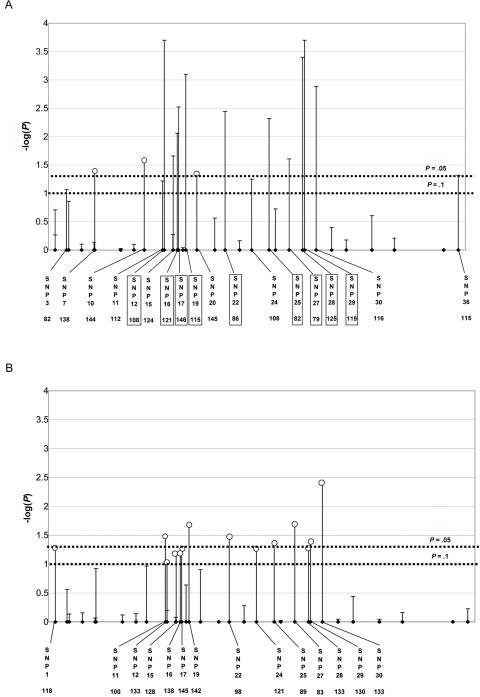
References
Web Resources
-
- Clarice R. Weinberg's Web site, http://dir.niehs.nih.gov/dirbb/weinberg/weinberg.htm (for software for the triad multimarker [TRIMM] test)
-
- HapMap, http://www.hapmap.org
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for NAT2, RFC1, POLI, CASP9, and IRF6)
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
