

The nuclear factor kappaB-activator gene PLEKHG5 is mutated in a form of autosomal recessive lower motor neuron disease with childhood onset
- PMID: 17564964
- PMCID: PMC1950913
- DOI: 10.1086/518900
The nuclear factor kappaB-activator gene PLEKHG5 is mutated in a form of autosomal recessive lower motor neuron disease with childhood onset
Abstract
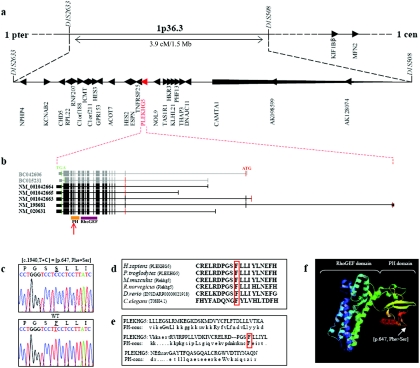
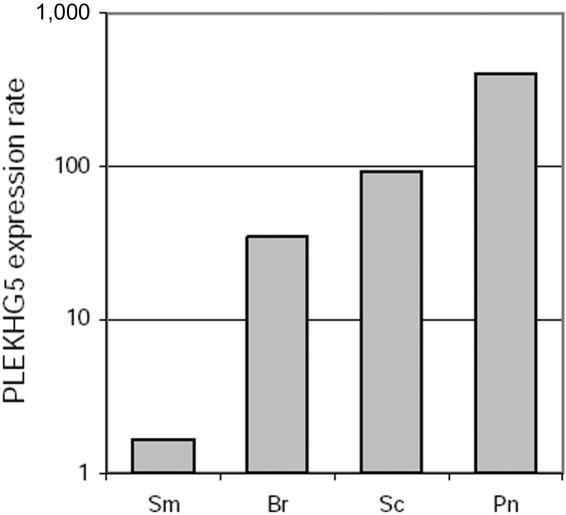
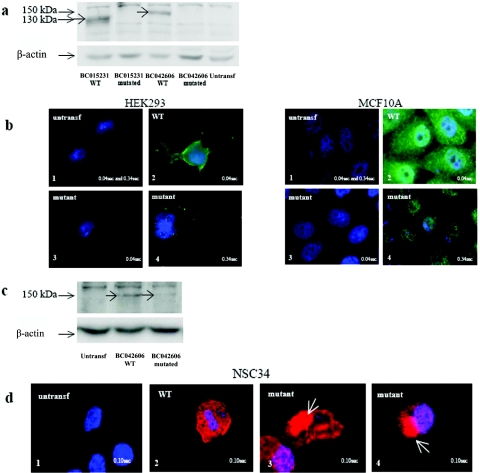
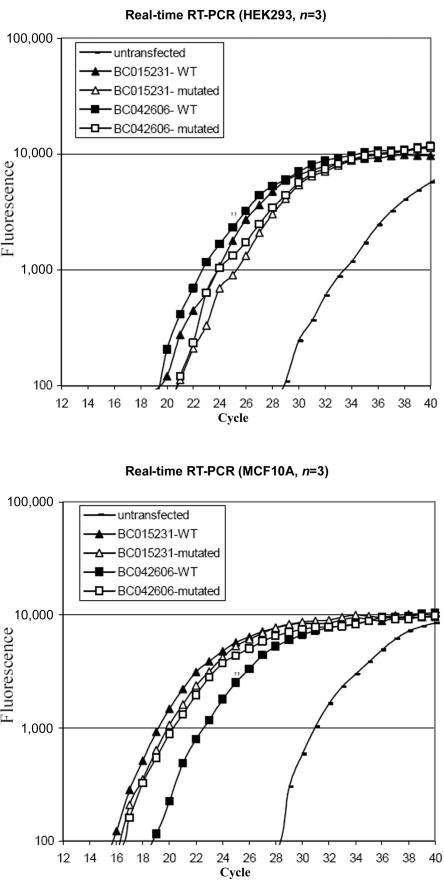
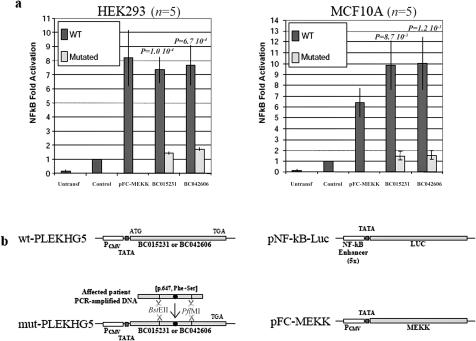
Lower motor neuron diseases (LMNDs) include a large spectrum of clinically and genetically heterogeneous disorders. Studying a large inbred African family, we recently described a novel autosomal recessive LMND variant characterized by childhood onset, generalized muscle involvement, and severe outcome, and we mapped the disease gene to a 3.9-cM interval on chromosome 1p36. We identified a homozygous missense mutation (c.1940 T-->C [p.647 Phe-->Ser]) of the Pleckstrin homology domain-containing, family G member 5 gene, PLEKHG5. In transiently transfected HEK293 and MCF10A cell lines, we found that wild-type PLEKHG5 activated the nuclear factor kappa B (NF kappa B) signaling pathway and that both the stability and the intracellular location of mutant PLEKHG5 protein were altered, severely impairing the NF kappa B transduction pathway. Moreover, aggregates were observed in transiently transfected NSC34 murine motor neurons overexpressing the mutant PLEKHG5 protein. Both loss of PLEKHG5 function and aggregate formation may contribute to neurotoxicity in this novel form of LMND.
Figures
References
Web Resources
-
- Expression Profile Viewer, http://www.ncbi.nlm.nih.gov/UniGene/ESTProfileViewer.cgi?uglist=Hs.284232
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for mRNAs [accession numbers NM_020631, NM_198681, NM_001042663, NM_001042664, NM_001042665, BC042606, and BC015231)
-
- ModBase, http://modbase.compbio.ucsf.edu/
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for spinal muscular atrophy, d-HMNII, d-HMNV, d-HMNVII, SMARD, DI-CMTB, centronuclear myopathy, ALS2, IAHSP, PLS, Kennedy disease, and amyotrophic lateral sclerosis)
References
-
- Grohmann K, Schuelke M, Diers A, Hoffmann K, Lucke B, Adams C, Bertini E, Leonhardt-Horti H, Muntoni F, Ouvrier R, et al (2001) Mutations in the gene encoding immunoglobulin μ-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet 29:75–7710.1038/ng703 - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
