

Cellular and clinical impact of haploinsufficiency for genes involved in ATR signaling
- PMID: 17564965
- PMCID: PMC1950915
- DOI: 10.1086/518696
Cellular and clinical impact of haploinsufficiency for genes involved in ATR signaling
Abstract
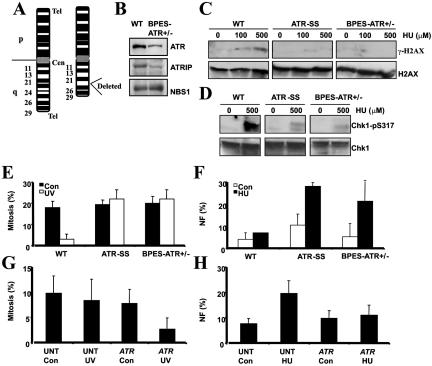
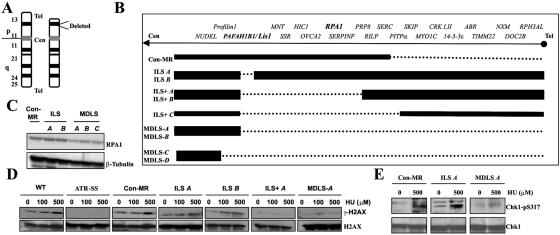
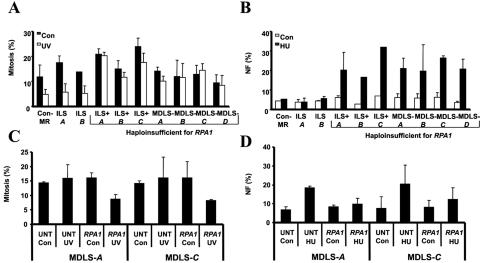
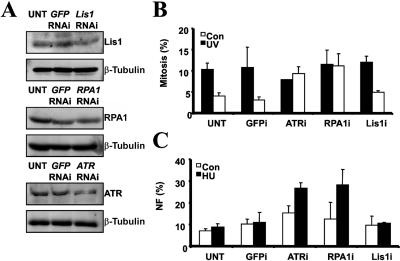
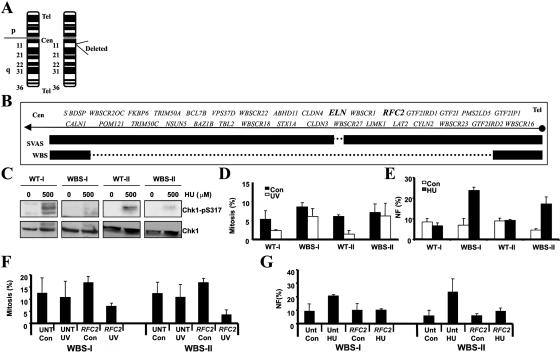
Ataxia telangiectasia and Rad3-related (ATR) protein, a kinase that regulates a DNA damage-response pathway, is mutated in ATR-Seckel syndrome (ATR-SS), a disorder characterized by severe microcephaly and growth delay. Impaired ATR signaling is also observed in cell lines from additional disorders characterized by microcephaly and growth delay, including non-ATR-SS, Nijmegen breakage syndrome, and MCPH1 (microcephaly, primary autosomal recessive, 1)-dependent primary microcephaly. Here, we examined ATR-pathway function in cell lines from three haploinsufficient contiguous gene-deletion disorders--a subset of blepharophimosis-ptosis-epicanthus inversus syndrome, Miller-Dieker lissencephaly syndrome, and Williams-Beuren syndrome--in which the deleted region encompasses ATR, RPA1, and RFC2, respectively. These three genes function in ATR signaling. Cell lines from these disorders displayed an impaired ATR-dependent DNA damage response. Thus, we describe ATR signaling as a pathway unusually sensitive to haploinsufficiency and identify three further human disorders displaying a defective ATR-dependent DNA damage response. The striking correlation of ATR-pathway dysfunction with the presence of microcephaly and growth delay strongly suggests a causal relationship.
Figures
References
Web Resource
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for ATR, SS, Nijmegen breakage syndrome, Fanconi anemia, MCPH1, BPES, FOXL2, PAHFAH1B1/Lis1, MDLS, RPA1, WBS, SVAS, ELN, and RFC2)
References
-
- O’Driscoll M, Jeggo PA (2003) Clinical impact of ATR checkpoint signalling failure in humans. Cell Cycle 2:194–195 - PubMed
-
- Seckel HPG (1960) Bird-headed dwarfs: studies in developmental anthropology including human proportions. Springer Karger, Basel, Switzerland
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
