

A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency
- PMID: 17564966
- PMCID: PMC1950923
- DOI: 10.1086/519219
A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency
Abstract
The acyl-CoA dehydrogenases are a family of multimeric flavoenzymes that catalyze the alpha,beta -dehydrogenation of acyl-CoA esters in fatty acid beta -oxidation and amino acid catabolism. Genetic defects have been identified in most of the acyl-CoA dehydrogenases in humans. Acyl-CoA dehydrogenase 9 (ACAD9) is a recently identified acyl-CoA dehydrogenase that demonstrates maximum activity with unsaturated long-chain acyl-CoAs. We now report three cases of ACAD9 deficiency. Patient 1 was a 14-year-old, previously healthy boy who died of a Reye-like episode and cerebellar stroke triggered by a mild viral illness and ingestion of aspirin. Patient 2 was a 10-year-old girl who first presented at age 4 mo with recurrent episodes of acute liver dysfunction and hypoglycemia, with otherwise minor illnesses. Patient 3 was a 4.5-year-old girl who died of cardiomyopathy and whose sibling also died of cardiomyopathy at age 21 mo. Mild chronic neurologic dysfunction was reported in all three patients. Defects in ACAD9 mRNA were identified in the first two patients, and all patients manifested marked defects in ACAD9 protein. Despite a significant overlap of substrate specificity, it appears that ACAD9 and very-long-chain acyl-CoA dehydrogenase are unable to compensate for each other in patients with either deficiency. Studies of the tissue distribution and gene regulation of ACAD9 and very-long-chain acyl-CoA dehydrogenase identify the presence of two independently regulated functional pathways for long-chain fat metabolism, indicating that these two enzymes are likely to be involved in different physiological functions.
Figures

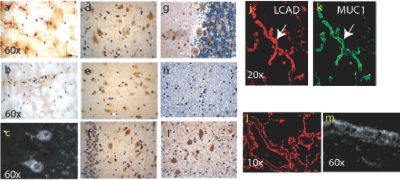
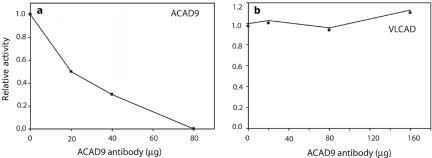


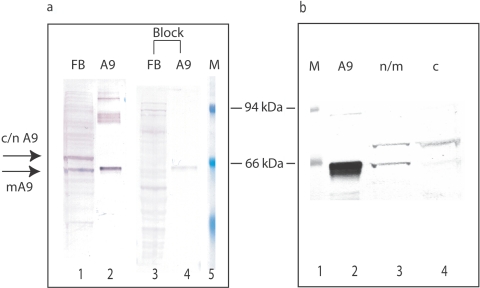


References
Web Resources
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for full-length human ACAD9 cDNA sequence from mRNA variant b [accession number NM_014049] and partial human ACAD9 cDNA sequence from mRNA variant a [accession numbers CR613592 and BX415793])
References
-
- Furuta S, Miyazawa S, Hashimoto T (1981) Purification and properties of rat liver acyl-CoA dehydrogenases and electron transfer flavoprotein. J Biochem (Tokyo) 90:1739–1750 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
