

Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa
- PMID: 17564971
- PMCID: PMC1950922
- DOI: 10.1086/518426
Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa
Abstract
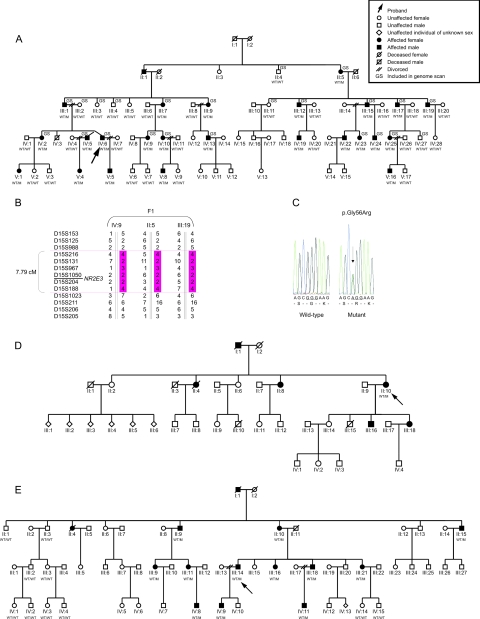
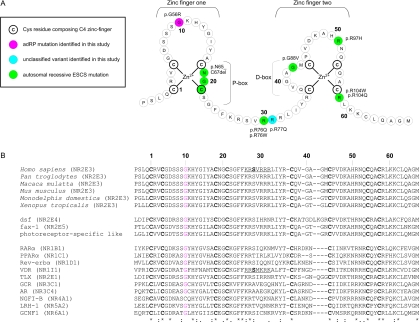
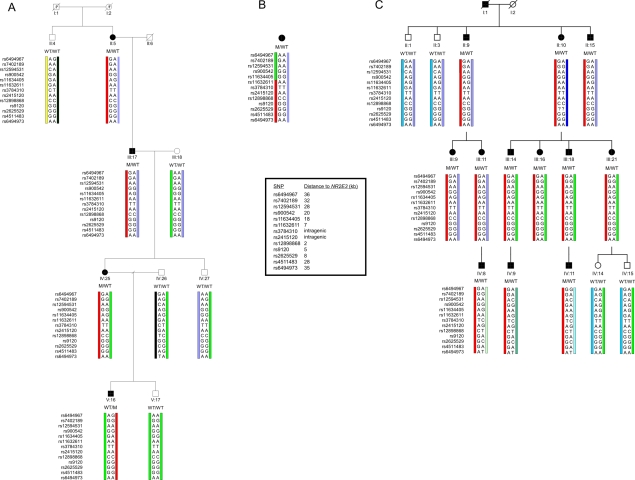
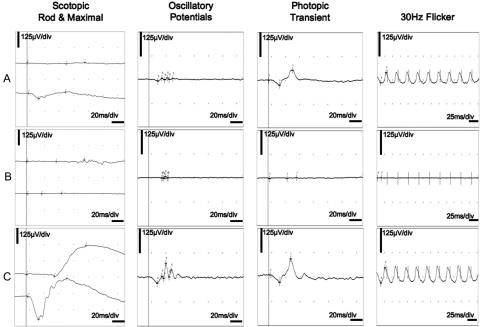
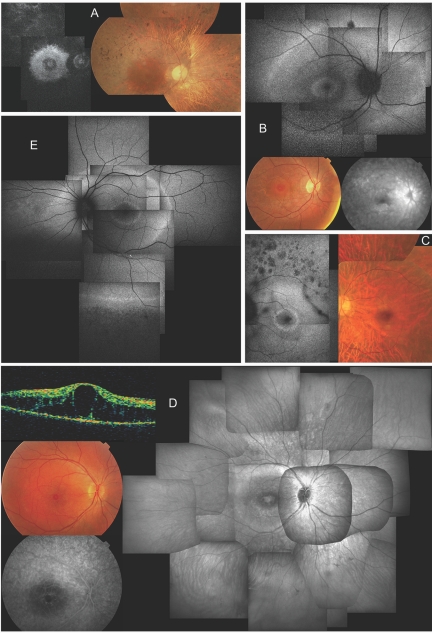
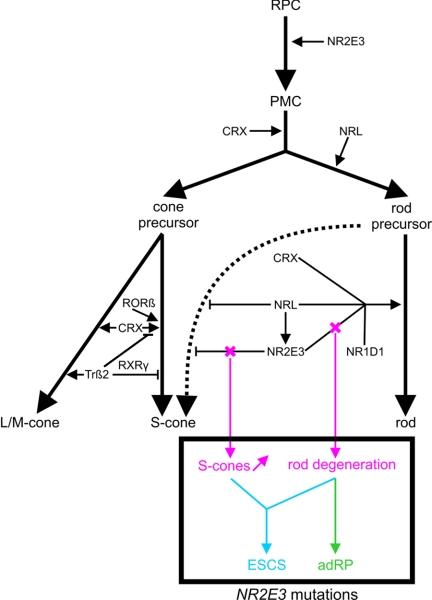
"Autosomal dominant retinitis pigmentosa" (adRP) refers to a genetically heterogeneous group of retinal dystrophies, in which 54% of all cases can be attributed to 17 disease loci. Here, we describe the localization and identification of the photoreceptor cell-specific nuclear receptor gene NR2E3 as a novel disease locus and gene for adRP. A heterozygous mutation c.166G-->A (p.Gly56Arg) was identified in the first zinc finger of NR2E3 in a large Belgian family affected with adRP. Overall, this missense mutation was found in 3 families affected with adRP among 87 unrelated families with potentially dominant retinal dystrophies (3.4%), of which 47 were affected with RP (6.4%). Interestingly, affected members of these families display a novel recognizable NR2E3-related clinical subtype of adRP. Other mutations of NR2E3 have previously been shown to cause autosomal recessive enhanced S-cone syndrome, a specific retinal phenotype. We propose a different pathogenetic mechanism for these distinct dominant and recessive phenotypes, which may be attributed to the dual key role of NR2E3 in the regulation of photoreceptor-specific genes during rod development and maintenance.
Figures
References
Web Resources
-
- Berkeley Drosophila Genome Project (BDGP), http://www.fruitfly.org/seq_tools/splice.html
-
- Ensembl Genome Browser, http://www.ensembl.org/index.html
-
- ESEfinder, http://rulai.cshl.edu/tools/ESE/
-
- International HapMap Project, http://www.hapmap.org/
-
- NCBI Map Viewer, http://www.ncbi.nlm.nih.gov/mapview/static/MVstart.html
References
-
- Bird AC (1995) Retinal photoreceptor dystrophies LI. Am J Ophthalmol 119:543–562 - PubMed
-
- Sullivan LS, Bowne SJ, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Lewis RA, Garcia CA, Ruiz RS, Blanton SH, Northrup H, et al (2006) Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci 47:3052–306410.1167/iovs.05-1443 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
