

Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome
- PMID: 17564974
- PMCID: PMC1950929
- DOI: 10.1086/519494
Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome
Abstract
Meckel syndrome (MKS) is a rare autosomal recessive lethal condition characterized by central nervous system malformations, polydactyly, multicystic kidney dysplasia, and ductal changes of the liver. Three loci have been mapped (MKS1-MKS3), and two genes have been identified (MKS1/FLJ20345 and MKS3/TMEM67), whereas the gene at the MKS2 locus remains unknown. To identify new MKS loci, a genomewide linkage scan was performed using 10-cM-resolution microsatellite markers in eight families. The highest heterogeneity LOD score was obtained for chromosome 12, in an interval containing CEP290, a gene recently identified as causative of Joubert syndrome (JS) and isolated Leber congenital amaurosis. In view of our recent findings of allelism, at the MKS3 locus, between these two disorders, CEP290 was considered a candidate, and homozygous or compound heterozygous truncating mutations were identified in four families. Sequencing of additional cases identified CEP290 mutations in two fetuses with MKS and in four families presenting a cerebro-reno-digital syndrome, with a phenotype overlapping MKS and JS, further demonstrating that MKS and JS can be variable expressions of the same ciliopathy. These data identify a fourth locus for MKS (MKS4) and the CEP290 gene as responsible for MKS.
Figures
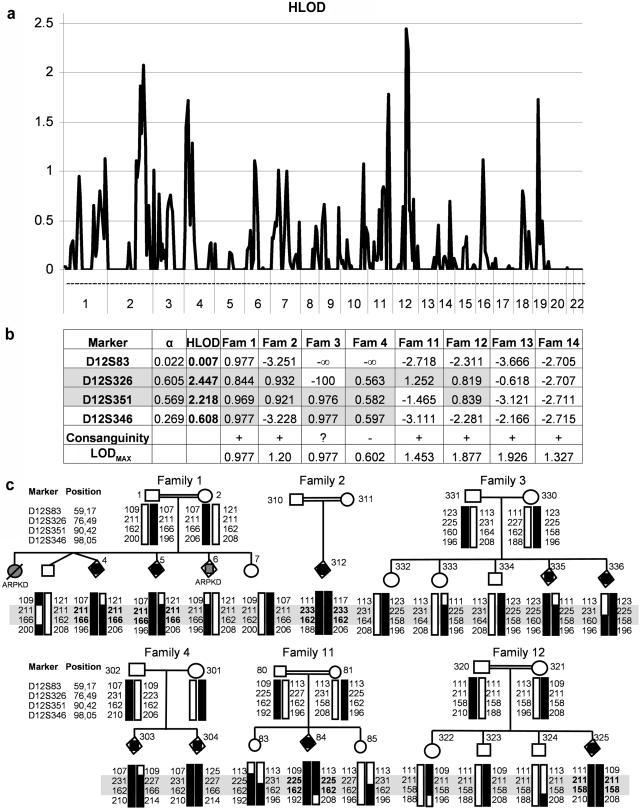
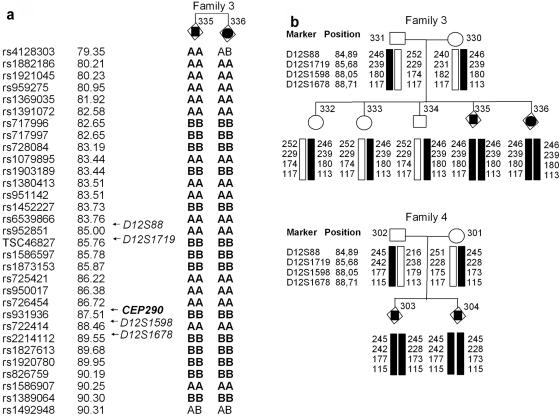
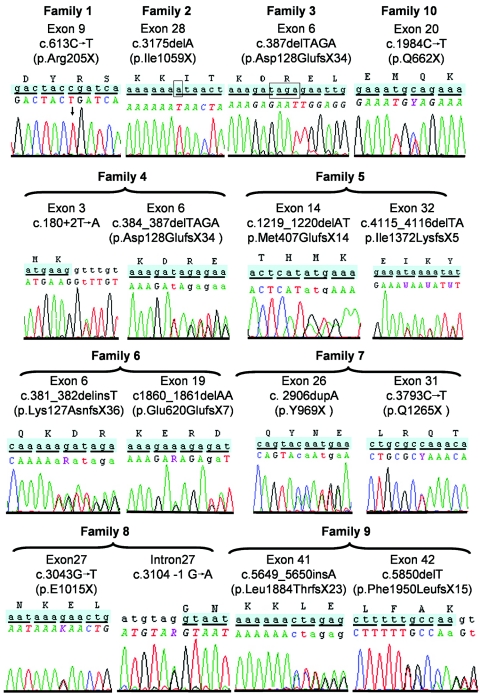
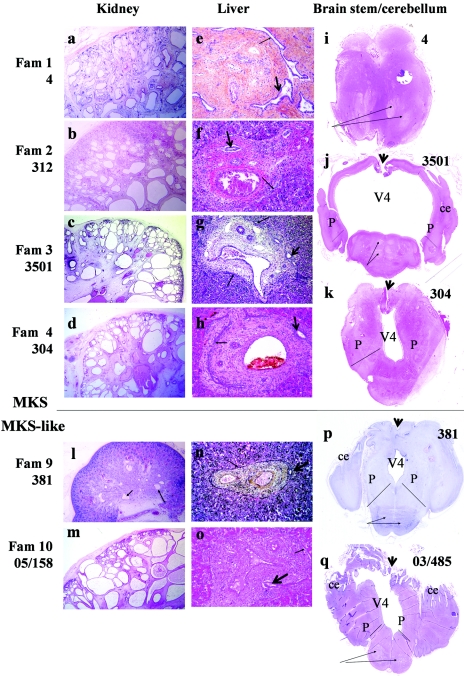
References
Web Resources
-
- GenBank, http://ncbi.nlm.nih.gov/Genbank/ (for cDNA translation codon [accession number NM_025114.3])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MKS, Meckel-like/Goldston syndrome, JS, LCA, and BBS)
-
- UCSC Genome Browser, http://genome.ucsc.edu/
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
