

Glycosylation, transport, and complex formation of palmitoyl protein thioesterase 1 (PPT1)--distinct characteristics in neurons
- PMID: 17565660
- PMCID: PMC1906764
- DOI: 10.1186/1471-2121-8-22
Glycosylation, transport, and complex formation of palmitoyl protein thioesterase 1 (PPT1)--distinct characteristics in neurons
Abstract
Background: Neuronal ceroid lipofuscinoses (NCLs) are collectively the most common type of recessively inherited childhood encephalopathies. The most severe form of NCL, infantile neuronal ceroid lipofuscinosis (INCL), is caused by mutations in the CLN1 gene, resulting in a deficiency of the lysosomal enzyme, palmitoyl protein thioesterase 1 (PPT1). The deficiency of PPT1 causes a specific death of neocortical neurons by a mechanism, which is currently unclear. To understand the function of PPT1 in more detail, we have further analyzed the basic properties of the protein, especially focusing on possible differences in non-neuronal and neuronal cells.
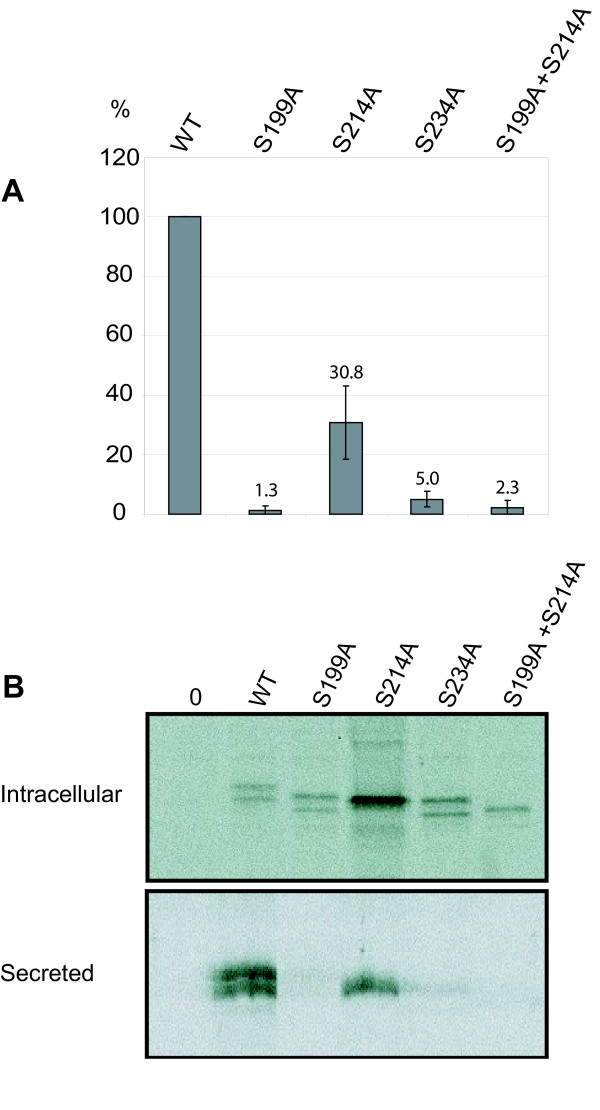
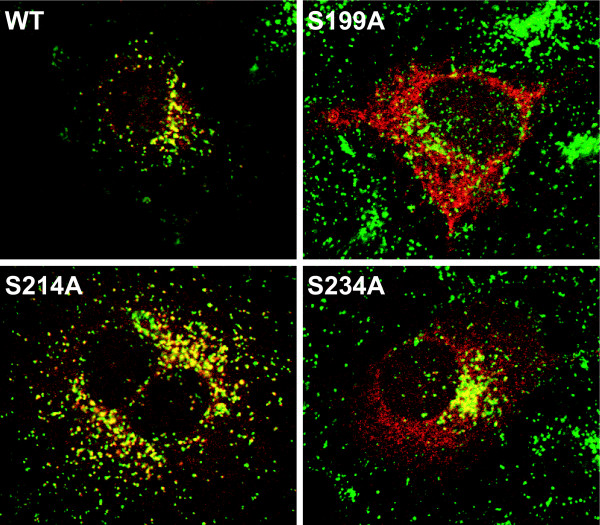
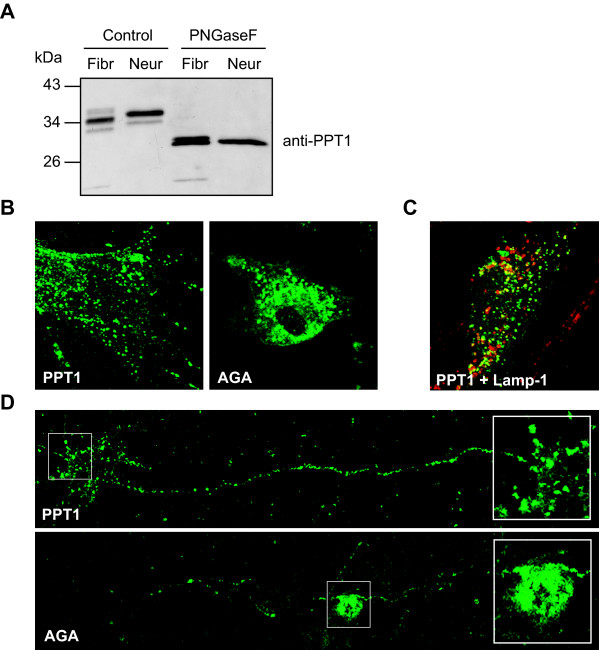
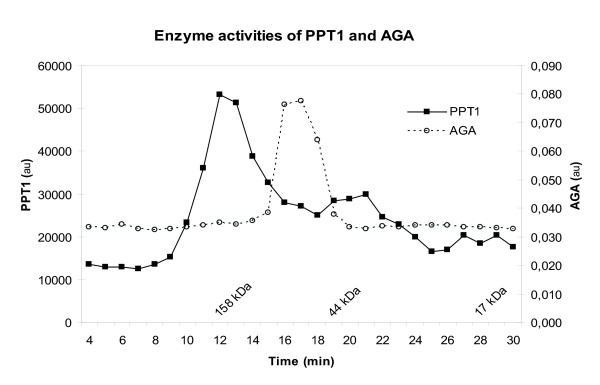
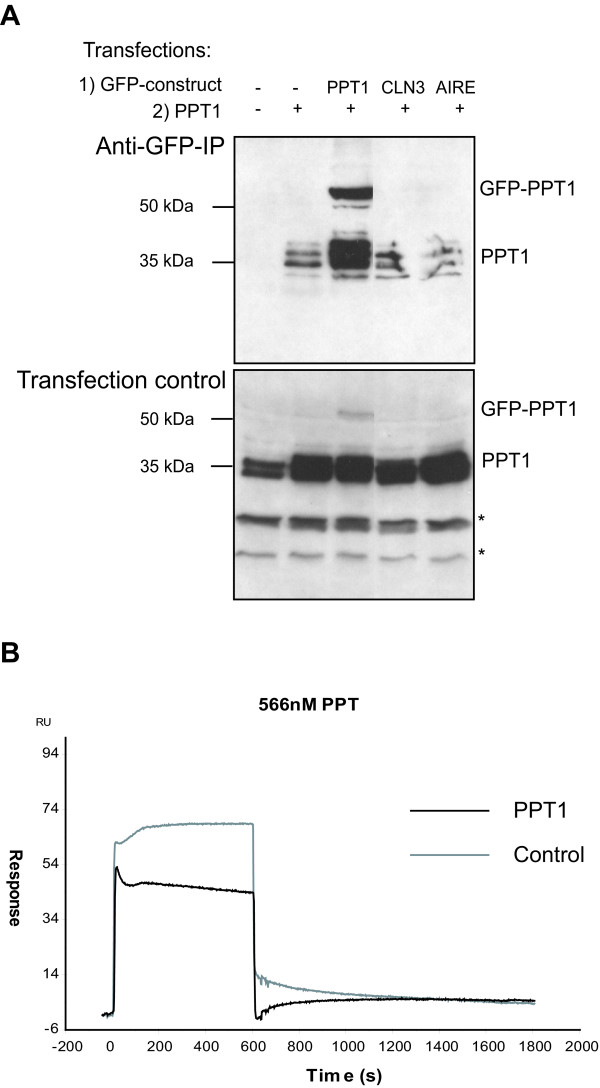
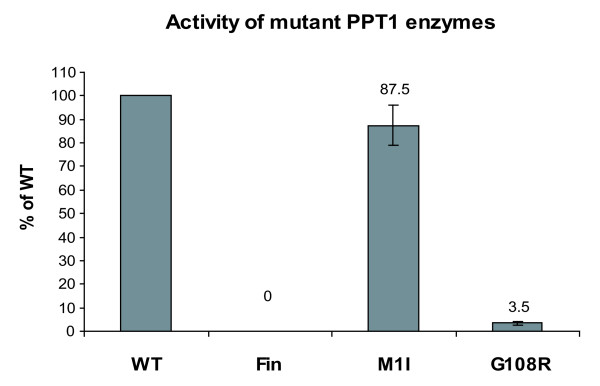
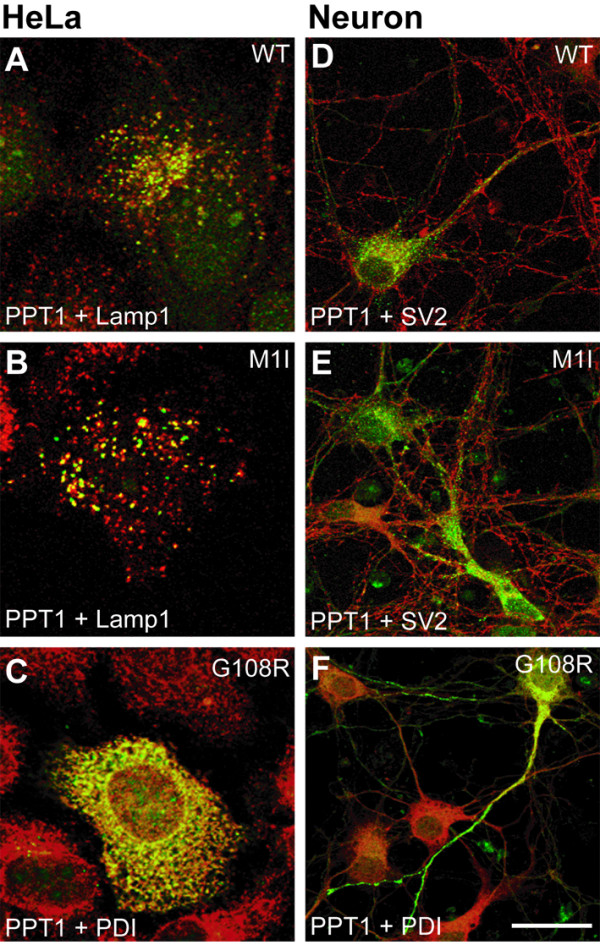
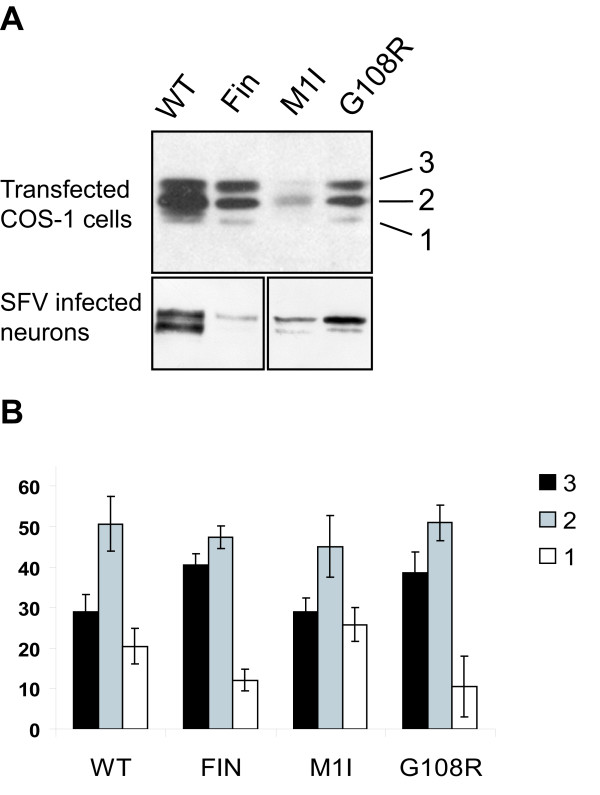
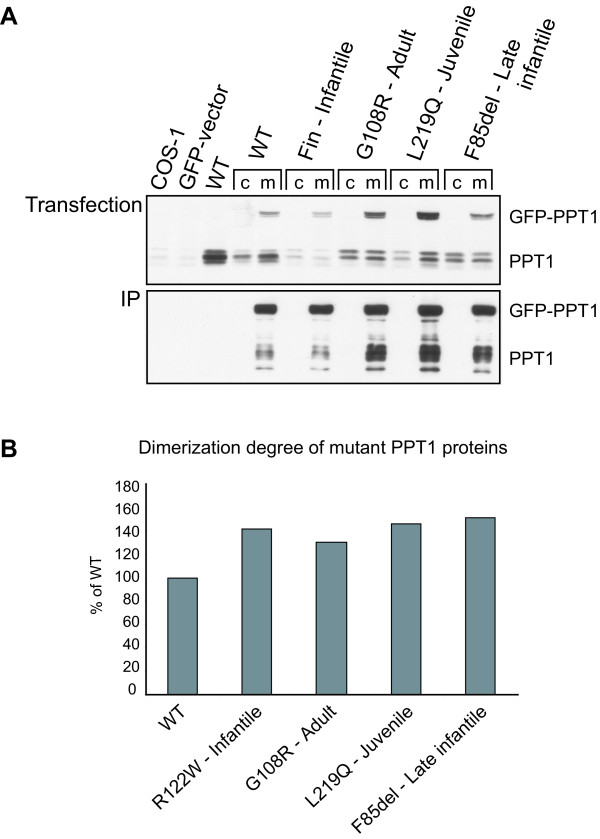
Results: Our study shows that the N-glycosylation of N197 and N232, but not N212, is essential for PPT1's activity and intracellular transport. Deglycosylation of overexpressed PPT1 produced in neurons and fibroblasts demonstrates differentially modified PPT1 in different cell types. Furthermore, antibody internalization assays showed differences in PPT1 transport when compared with a thoroughly characterized lysosomal enzyme aspartylglucosaminidase (AGA), an important observation potentially influencing therapeutic strategies. PPT1 was also demonstrated to form oligomers by size-exclusion chromatography and co-immunoprecipitation assays. Finally, the consequences of disease mutations were analyzed in the perspective of our new results, suggesting that the mutations increase both the degree of glycosylation of PPT1 and its ability to form complexes.
Conclusion: Our current study describes novel properties for PPT1. We observe differences in PPT1 processing and trafficking in neuronal and non-neuronal cells, and describe for the first time the ability of PPT1 to form complexes. Understanding the basic characteristics of PPT1 is fundamental in order to clarify the molecular pathogenesis behind neurodegeneration in INCL.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
