

Modulation of proteinase K-resistant prion protein in cells and infectious brain homogenate by redox iron: implications for prion replication and disease pathogenesis
- PMID: 17567949
- PMCID: PMC1951779
- DOI: 10.1091/mbc.e07-04-0317
Modulation of proteinase K-resistant prion protein in cells and infectious brain homogenate by redox iron: implications for prion replication and disease pathogenesis
Abstract
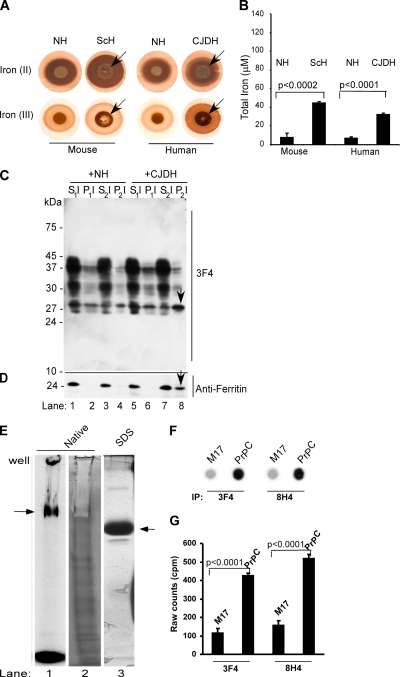
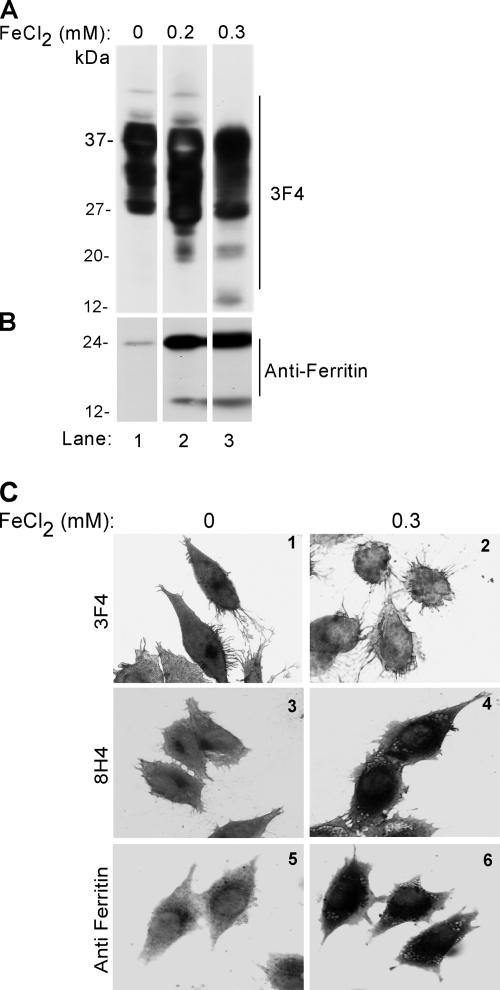
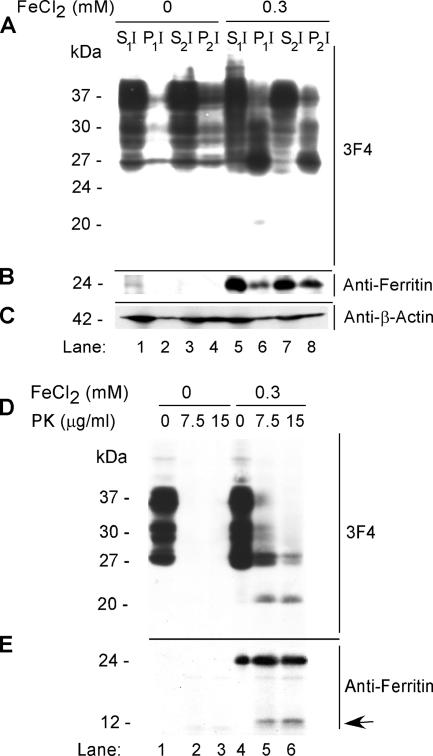
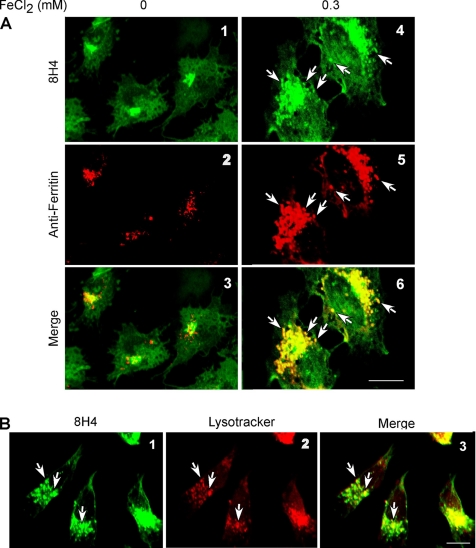
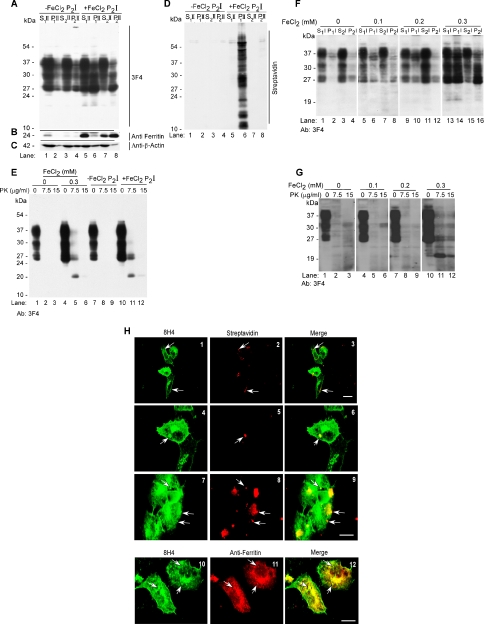
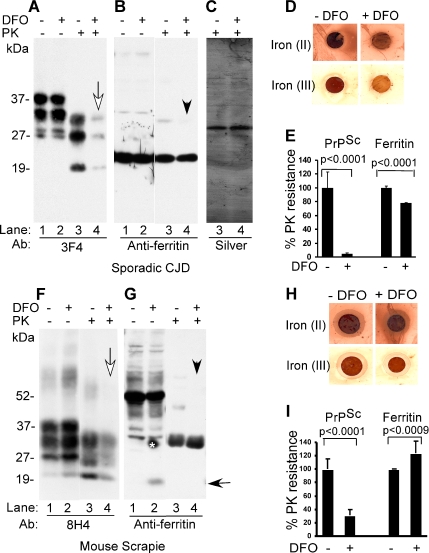
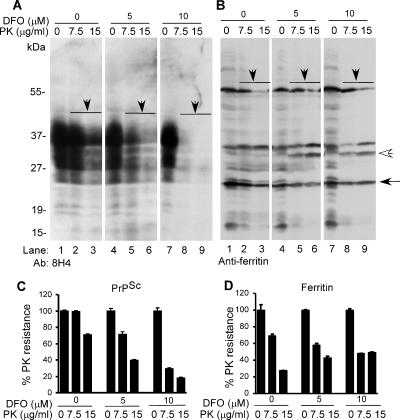
The principal infectious and pathogenic agent in all prion disorders is a beta-sheet-rich isoform of the cellular prion protein (PrP(C)) termed PrP-scrapie (PrP(Sc)). Once initiated, PrP(Sc) is self-replicating and toxic to neuronal cells, but the underlying mechanisms remain unclear. In this report, we demonstrate that PrP(C) binds iron and transforms to a PrP(Sc)-like form (*PrP(Sc)) when human neuroblastoma cells are exposed to an inorganic source of redox iron. The *PrP(Sc) thus generated is itself redox active, and it induces the transformation of additional PrP(C), simulating *PrP(Sc) propagation in the absence of brain-derived PrP(Sc). Moreover, limited depletion of iron from prion disease-affected human and mouse brain homogenates and scrapie-infected mouse neuroblastoma cells results in 4- to 10-fold reduction in proteinase K (PK)-resistant PrP(Sc), implicating redox iron in the generation, propagation, and stability of PK-resistant PrP(Sc). Furthermore, we demonstrate increased redox-active ferrous iron levels in prion disease-affected brains, suggesting that accumulation of PrP(Sc) is modulated by the combined effect of imbalance in brain iron homeostasis and the redox-active nature of PrP(Sc). These data provide information on the mechanism of replication and toxicity by PrP(Sc), and they evoke predictable and therapeutically amenable ways of modulating PrP(Sc) load.
Figures
References
-
- Aguzzi A., Polymendiou M. Mammalian prion biology: one century of evolving concepts. Cell. 2004;116:313–327. - PubMed
-
- Andreoletti O., Levavasseur E., Uro-Coste E., Tabouret G., Sarradin P., Delisle M. B., Berthon P., Salvayre R., Schelcher F., Negre-Salvayre A. Astrocytes accumulate 4-hydroxynonenal adducts in murine scrapie and human Creutzfeldt-Jakob disease. Neurobiol. Dis. 2002;11:386–393. - PubMed
-
- Behl C., Davis J. B., Lesley R., Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. - PubMed
-
- Brazier M. W., et al. Correlative studies support lipid peroxidation is linked to PrP(res) propagation as an early primary pathogenic event in prion disease. Brain Res. Bull. 2006;68:346–354. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
