

Prominent use of distal 5' transcription start sites and discovery of a large number of additional exons in ENCODE regions
- PMID: 17567994
- PMCID: PMC1891335
- DOI: 10.1101/gr.5660607
Prominent use of distal 5' transcription start sites and discovery of a large number of additional exons in ENCODE regions
Abstract
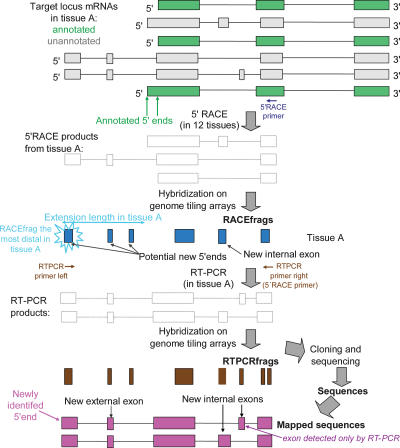
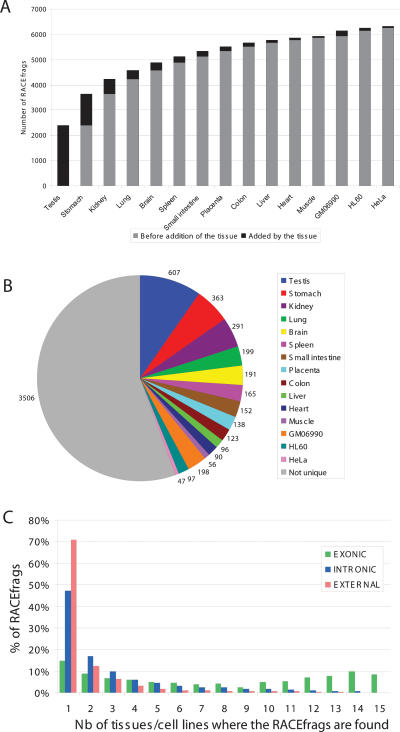
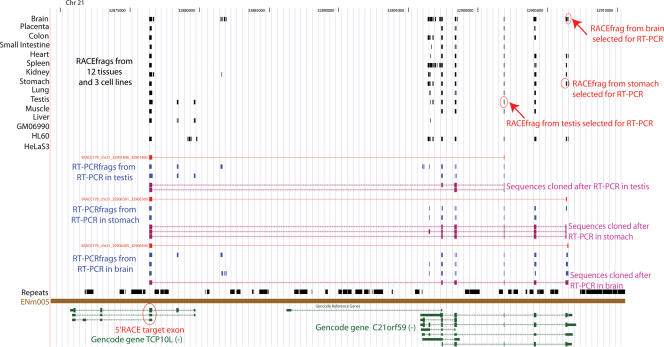
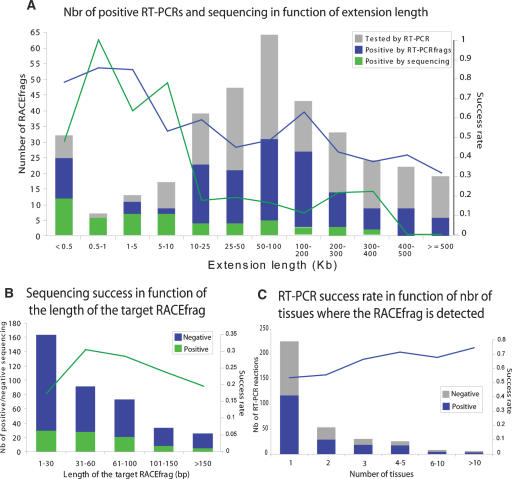
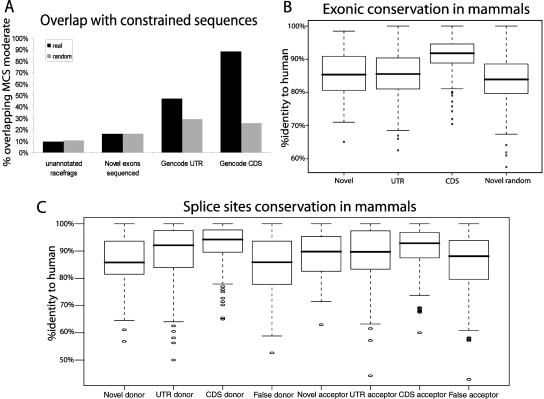
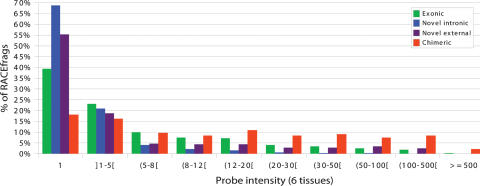
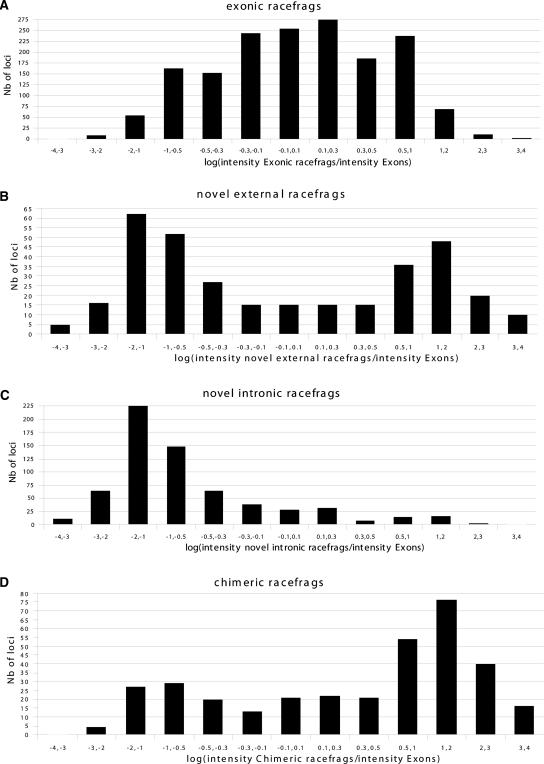
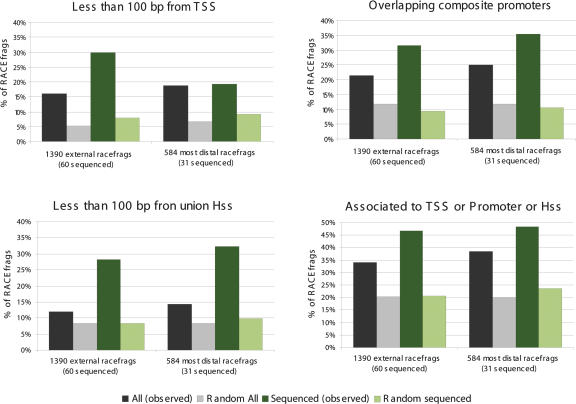
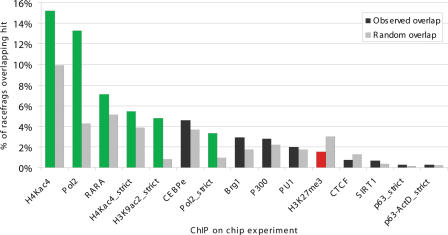
This report presents systematic empirical annotation of transcript products from 399 annotated protein-coding loci across the 1% of the human genome targeted by the Encyclopedia of DNA elements (ENCODE) pilot project using a combination of 5' rapid amplification of cDNA ends (RACE) and high-density resolution tiling arrays. We identified previously unannotated and often tissue- or cell-line-specific transcribed fragments (RACEfrags), both 5' distal to the annotated 5' terminus and internal to the annotated gene bounds for the vast majority (81.5%) of the tested genes. Half of the distal RACEfrags span large segments of genomic sequences away from the main portion of the coding transcript and often overlap with the upstream-annotated gene(s). Notably, at least 20% of the resultant novel transcripts have changes in their open reading frames (ORFs), most of them fusing ORFs of adjacent transcripts. A significant fraction of distal RACEfrags show expression levels comparable to those of known exons of the same locus, suggesting that they are not part of very minority splice forms. These results have significant implications concerning (1) our current understanding of the architecture of protein-coding genes; (2) our views on locations of regulatory regions in the genome; and (3) the interpretation of sequence polymorphisms mapping to regions hitherto considered to be "noncoding," ultimately relating to the identification of disease-related sequence alterations.
Figures
References
-
- Adams M.D., Celniker S.E., Holt R.A., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Celniker S.E., Holt R.A., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Holt R.A., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Li P.W., Hoskins R.A., Galle R.F., Hoskins R.A., Galle R.F., Galle R.F., et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. - PubMed
-
- Akiva P., Toporik A., Edelheit S., Peretz Y., Diber A., Shemesh R., Novik A., Sorek R., Toporik A., Edelheit S., Peretz Y., Diber A., Shemesh R., Novik A., Sorek R., Edelheit S., Peretz Y., Diber A., Shemesh R., Novik A., Sorek R., Peretz Y., Diber A., Shemesh R., Novik A., Sorek R., Diber A., Shemesh R., Novik A., Sorek R., Shemesh R., Novik A., Sorek R., Novik A., Sorek R., Sorek R. Transcription-mediated gene fusion in the human genome. Genome Res. 2006;16:30–36. - PMC - PubMed
-
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Dolinski K., Dwight S.S., Eppig J.T., Dwight S.S., Eppig J.T., Eppig J.T., et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. - PMC - PubMed
-
- Bertone P., Stolc V., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Stolc V., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Royce T.E., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Rozowsky J.S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Urban A.E., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Zhu X., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Rinn J.L., Tongprasit W., Samanta M., Weissman S., Tongprasit W., Samanta M., Weissman S., Samanta M., Weissman S., Weissman S., et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials