

Analyses of deep mammalian sequence alignments and constraint predictions for 1% of the human genome
- PMID: 17567995
- PMCID: PMC1891336
- DOI: 10.1101/gr.6034307
Analyses of deep mammalian sequence alignments and constraint predictions for 1% of the human genome
Abstract
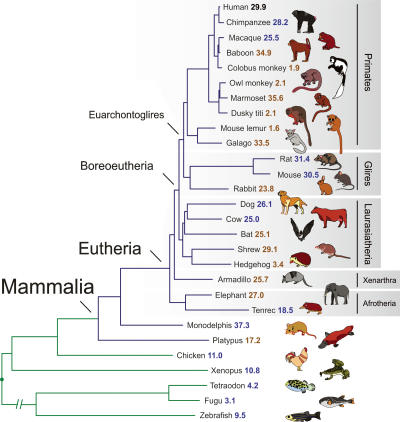
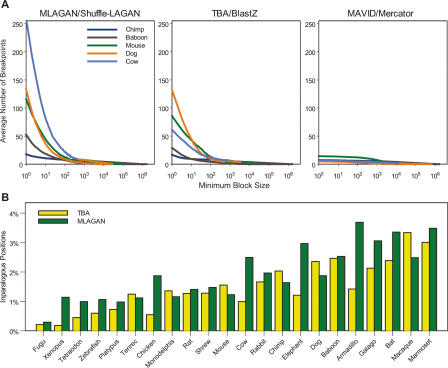
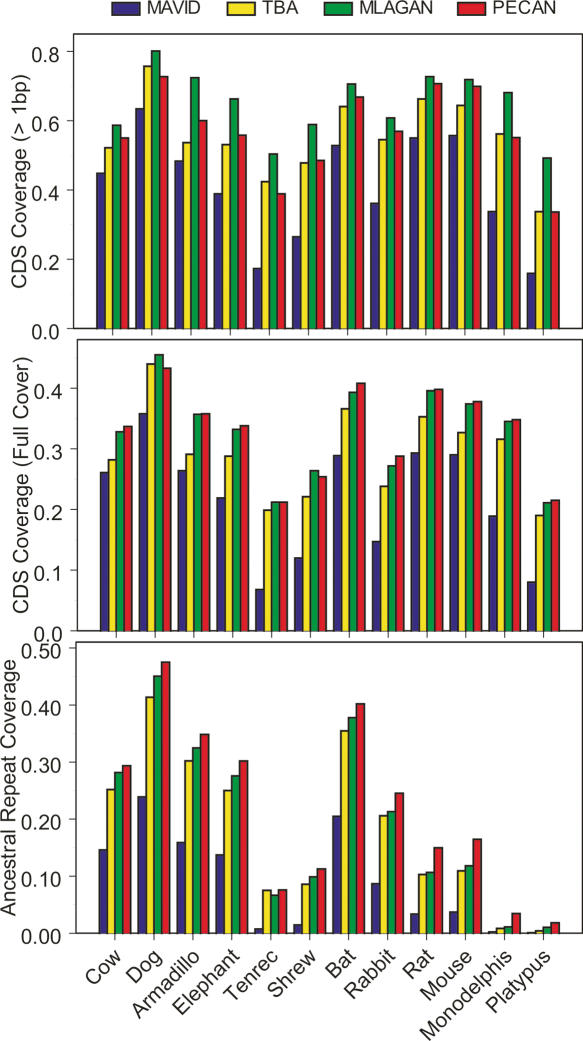
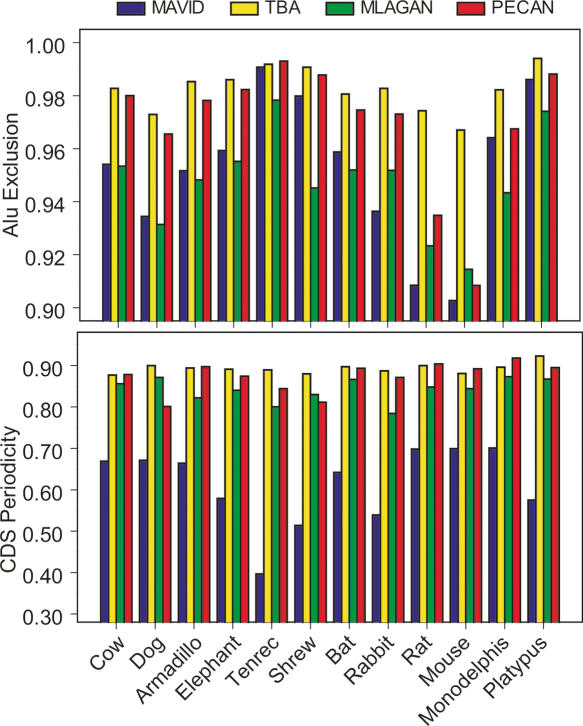
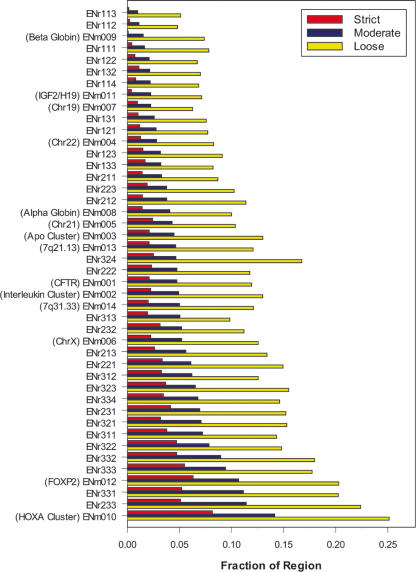
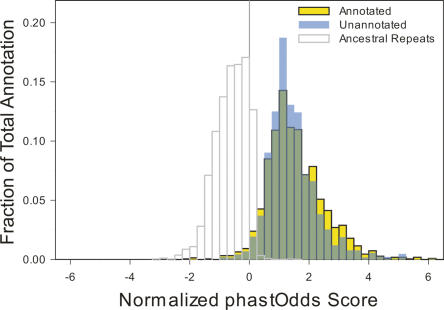
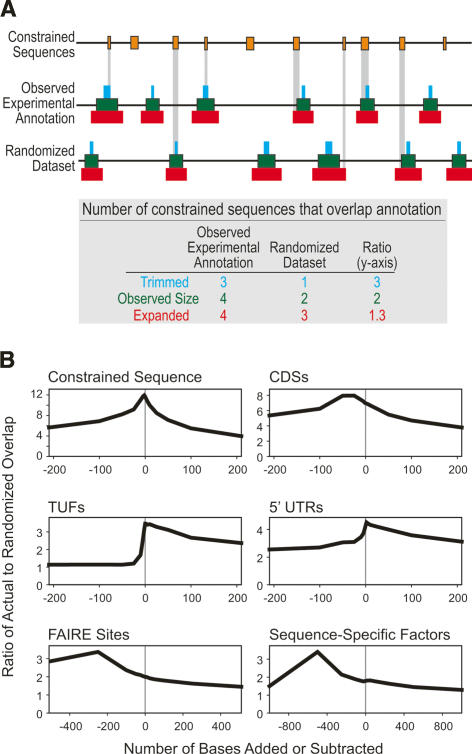
A key component of the ongoing ENCODE project involves rigorous comparative sequence analyses for the initially targeted 1% of the human genome. Here, we present orthologous sequence generation, alignment, and evolutionary constraint analyses of 23 mammalian species for all ENCODE targets. Alignments were generated using four different methods; comparisons of these methods reveal large-scale consistency but substantial differences in terms of small genomic rearrangements, sensitivity (sequence coverage), and specificity (alignment accuracy). We describe the quantitative and qualitative trade-offs concomitant with alignment method choice and the levels of technical error that need to be accounted for in applications that require multisequence alignments. Using the generated alignments, we identified constrained regions using three different methods. While the different constraint-detecting methods are in general agreement, there are important discrepancies relating to both the underlying alignments and the specific algorithms. However, by integrating the results across the alignments and constraint-detecting methods, we produced constraint annotations that were found to be robust based on multiple independent measures. Analyses of these annotations illustrate that most classes of experimentally annotated functional elements are enriched for constrained sequences; however, large portions of each class (with the exception of protein-coding sequences) do not overlap constrained regions. The latter elements might not be under primary sequence constraint, might not be constrained across all mammals, or might have expendable molecular functions. Conversely, 40% of the constrained sequences do not overlap any of the functional elements that have been experimentally identified. Together, these findings demonstrate and quantify how many genomic functional elements await basic molecular characterization.
Figures
References
-
- Aparicio S., Chapman J., Stupka E., Putnam N., Chia J.-M., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Chapman J., Stupka E., Putnam N., Chia J.-M., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Stupka E., Putnam N., Chia J.-M., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Putnam N., Chia J.-M., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Chia J.-M., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Christoffels A., Rash S., Hoon S., Smit A., Rash S., Hoon S., Smit A., Hoon S., Smit A., Smit A., et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–1310. - PubMed
-
- Blakesley R.W., Hansen N.F., Mullikin J.C., Thomas P.J., McDowell J.C., Maskeri B., Young A.C., Benjamin B., Brooks S.Y., Coleman B.I., Hansen N.F., Mullikin J.C., Thomas P.J., McDowell J.C., Maskeri B., Young A.C., Benjamin B., Brooks S.Y., Coleman B.I., Mullikin J.C., Thomas P.J., McDowell J.C., Maskeri B., Young A.C., Benjamin B., Brooks S.Y., Coleman B.I., Thomas P.J., McDowell J.C., Maskeri B., Young A.C., Benjamin B., Brooks S.Y., Coleman B.I., McDowell J.C., Maskeri B., Young A.C., Benjamin B., Brooks S.Y., Coleman B.I., Maskeri B., Young A.C., Benjamin B., Brooks S.Y., Coleman B.I., Young A.C., Benjamin B., Brooks S.Y., Coleman B.I., Benjamin B., Brooks S.Y., Coleman B.I., Brooks S.Y., Coleman B.I., Coleman B.I., et al. An intermediate grade of finished genomic sequence suitable for comparative analyses. Genome Res. 2004;14:2235–2244. - PMC - PubMed
-
- Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Kent W.J., Riemer C., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Riemer C., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Elnitski L., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Smit A.F.A., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Rosenbloom K., Clawson H., Green E.D., Clawson H., Green E.D., Green E.D., et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. - PMC - PubMed
-
- Boffelli D., McAuliffe J., Ovcharenko D., Lewis K.D., Ovcharenko I., Pachter L., Rubin E.M., McAuliffe J., Ovcharenko D., Lewis K.D., Ovcharenko I., Pachter L., Rubin E.M., Ovcharenko D., Lewis K.D., Ovcharenko I., Pachter L., Rubin E.M., Lewis K.D., Ovcharenko I., Pachter L., Rubin E.M., Ovcharenko I., Pachter L., Rubin E.M., Pachter L., Rubin E.M., Rubin E.M. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003;299:1391–1394. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources